KOMBINIERTE ANALYSE EINES AKTUALISIERTEN SPEKTRUMS AN PESTIZIDEN IN TRINKWASSER MITTELS HOCHLEISTUNGSFLÜSSIGKEITSCHROMATGRAPHIE/ DIODENARRAYDETEKTION UND GASCHROMATOGRAPHIE MIT PROGR.TEMP.VAPORIZ.
Aus der Abteilung III - Lebensmittelchemie/Ökochemie (Abteilungsleiter: Oberfeldapotheker Dr. M. Gerhartz) des Zentralen Instituts des Sanitätsdienstes der Bundeswehr Koblenz (Leiter: Flottenarzt Dr. H. Bergmann)
von Markus Küsters, Rainer Gallitzendörfer, Thomas Timm, Dagmar Koch, Sebastian Reeser, Ute Bimber und Michael Gerhartz
Zusammenfassung:
Hintergrund: Pestizide besitzen eine große Bedeutung im Pflanzenschutz und werden weltweit angewendet.
Unter den Pestiziden nehmen Herbizide und Fungizide eine besondere Stellung ein. Durch die Verwendung in der Landwirtschaft werden entsprechende Stoffmengen in Oberflächen-, Grund- und Trinkwasser eingetragen. Gemäß der Trinkwasserverordnung 2001 sind Pestizide und deren relevante Metaboliten daher regelmäßig in Trinkwasser zu überwachen.
Im Rahmen der Eigenvollzugskompetenz der Bundeswehr bei der Überwachung von Trinkwasser kommt daher dem Nachweis dieser Substanzen im Zuge des vorbeugenden Gesundheitsschutzes sowohl im Inland wie auch im Auslandseinsatz eine besondere Bedeutung zu. Um Untersuchungsverfahren nach dem aktuellen wissenschaftlichen Stand vorhalten zu können, war deshalb eine Aktualisierung des Untersuchungsspektrums sowie der eingesetzten Analysenmethoden für die Untersuchung und Überwachung von Trinkwasser erforderlich.
Methoden:
Aufgrund der Vielzahl von Wirkstoffen wurde eine Priorisierung bezüglich der Anwendungsmengen und -bereiche vorgenommen. Von 252 in Deutschland zugelassenen Wirkstoffen wurden 111 mit einer besonderen Untersuchungsrelevanz identifiziert. Für 54 dieser Substanzen wurden Methoden zum Nachweis in Trinkwasser entwickelt. Dabei wurden die Analyten nach Festphasenextraktion aus Trinkwasser mittels Hochleistungsflüssigkeitschromatographie/Diodenarraydetektion (HPLC/DAD) und Gaschromatographie/ Massenspektrometrie (GC/MS) bzw. GC/mikro- Elektroneneinfangdetektor (GC/µECD) analysiert.
Ergebnisse:
Nachweisgrenzen von ≤ 25 ng/L sowie gute und reproduzierbare Wiederfindungsraten wurden für alle Substanzen erreicht. Die angewendeten Methoden zur Analyse der Herbizide und Fungizide in Trinkwasser sowie die durchgeführte Methodenentwicklung und Validierung werden vorgestellt.
Schlussfolgerungen:
Durch die Kombination der Probenvorbereitung und der chromatographischen Analyse wurden präzise Methoden entwickelt, die ein repräsentatives und aktuelles Spektrum an Pestiziden analytisch zugänglich machen.
Summary:
Background: Pesticides, especially herbicides and fungicides, have much importance for crop protection presenting the group of pesticides with most prevalent use. Due to the application in agriculture, respective amounts can pollute surface, ground and drinking water. Therefore, contents of pesticides and their metabolites need to be controlled according to the Council Directive 98/83/EC on the quality of water intended for human consumption. Hence, within the scope of the enforcement of this Act by the Federal Armed Forces a special significance attaches to the determination of these substances in the course of preventive health protection at home as well as foreign assignment. Because of both the multitude of active ingredients and the necessity of analyzing a representative selection of pesticides, a prioritization and an update of the analytical methods was carried out regarding extent of application and scope.
Methods:
111 of 252 authorized plant protection agents were classified as priority water pollutants. Hence, analytical methods for determining 54 of these substances in drinking water were developed so far. The analytes were measured by means of HPLC/DAD and GC-MS/µECD after solid phase extraction, respectively.
Results:
Limits of detection < of 25 ng/L as well as satisfying and reproducible recoveries were obtained over all. The applied analytical methods associated with method development and validation for the determination of herbicides and fungicides in drinking water are presented.
Conclusions:
Briefly, as a result of the combination of sample preparation and chromatographic analysis accurate methods for determination of a representative and current selection of pesticides were established.
1. Einführung
Pestizide (Pflanzenschutzmittel) besitzen eine große Bedeutung in der Landwirtschaft und Produktion pflanzlicher Lebensmittel. Unterteilt werden sie in mehrere Kategorien gemäß ihrem Wirkspektrum, von denen im Wesentlichen vier Gruppen, die Insektizide, Herbizide, Fungizide und Wachstumsregler, aufgrund hoher Absatzmengen oder ökochemischer Eigenschaften für die umweltchemische Analytik von Bedeutung sind. Insbesondere für die Trinkwasseranalytik ist eine weitergehende Begrenzung des Untersuchungsspektrums durch die Vielzahl an zugelassenen Pflanzenschutzmittelwirkstoffen sowie deren Anwendungsgebiete angemessen.
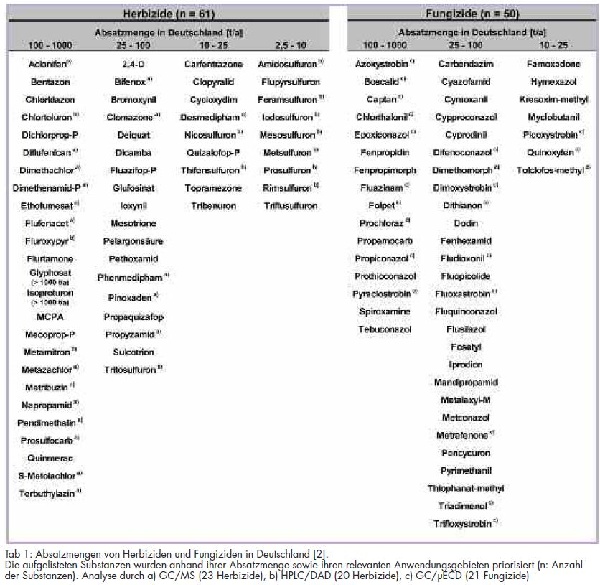
Derzeit sind in Deutschland 252 Wirkstoffe in 623 Pflanzenschutzmitteln mit 4 087 Anwendungen zugelassen. Durch die rechtlichen Vorgaben der Trinkwasserverordnung 2001 (TrinkWV 2001) [1] und auch aus ökotoxikologischen Betrachtungsgründen ist eine Überwachung von Trinkwasser auf Kontaminationen mit Pflanzenschutzmitteln zwingend erforderlich. Beiträge aus der Fachliteratur geben vermehrt Hinweise darauf, dass einige Wirkstoffe aus der Gruppe der Harnstoff-Herbizide aus dem Boden in das Grundwasser ausgewaschen werden können und dort eine hohe Persistenz aufweisen [2]. Für eine Einschätzung der Kontaminationswahrscheinlichkeit sind dabei neben dem ökologischen Verhalten und chemisch-physikalischen Eigenschaften der Absatz und das Anwendungsgebiet der einzelnen Substanzen zu berücksichtigen. Einen Überblick über den Absatz verschiedener Wirkstoffe in Deutschland gibt Tabelle 1.
Für die Überwachung von Trinkwasser nach den Vorgaben der Trinkwasserverordnung 2001 wurden aus den absatzstärksten Gruppen der Herbizide und Fungizide (53 760 bzw. 27 664 t/a) zugelassene Substanzen mit hohen Inlandsabsatzmengen [3] und weiten Anwendungsgebieten ausgewählt. Der Absatz an Insektiziden (einschließlich Akarizide und Pheromone) sowie Wachstumsregler (einschließlich Keimhemmungsmittel) lag zum Vergleich dazu im Jahr 2008 nur bei 6 557 und 7 217 t/a. So konnte eine Priorisierung von Wirkstoffen mit hohem Absatz unter gleichzeitiger Berücksichtigung der Verwendung in relevanten Einsatzgebieten, wie Ackerbau und Kulturen, vorgenommen werden. Hintergrund dieser Vorgehensweise war, dass viele in Standardverfahren beschriebene Analysenmethoden sowie Trinkwasserringversuche wenig gebräuchliche Herbizide analysieren, deren Anwendung oder Zulassung nicht mehr vorliegt und die weiterhin keine nennenswerten Persistenzen aufweisen. Eine Aktualisierung des Untersuchungsspektrums sowie der eingesetzten Analysenmethoden war somit erforderlich.
2. Methoden
2.1 Auswahl analytischer Methoden
Die Gruppe der Pflanzenschutzmittel beinhaltet eine Vielzahl unterschiedlichster Wirkstoffklassen, deren physikalisch-chemische Eigenschaften sehr different sind. Eine Unterscheidung der Substanzen muss demnach einerseits nach sauren, basischen oder neutralen funktionellen Gruppen und andererseits nach chromatographischen Kriterien, wie Polarität, Thermolabilität, Chromophore und Detektionsempfindlichkeit bezüglich Massenspektrometrie, Elektroneneinfang- oder optischer Detektion, erfolgen.
Die in die Methodenentwicklung einbezogenen Substanzen wurden nach den Kriterien der Thermolabilität und Polarität (HPLC) sowie nach sauren und basischen bzw. neutralen Eigenschaften ausgewählt. Durch die thermale Instabilität von Harnstoffderivaten ist eine gaschromatographische Analyse dieser Substanzen nicht möglich. Die Bestimmung mittels HPLC/DAD ist aber eine empfindliche und einfach durchzuführende Methode, die gleichzeitig auch eine Absicherung von positiven Befunden durch substanzspezifische UV-Spektren ermöglicht. Substanzen mit vielen Halogensubstitutionen können mit hoher Empfindlichkeit mittels eines Mikro-Elektroneneinfangdetektors (µECD) detektiert werden. Der µECD ist damit eine gute Alternative zur massenselektiven Detektion, die am häufigsten Anwendung findet, jedoch gelegentlich durch starke oder wenig spezifische Fragmentierungsreaktionen für die relevanten Analyten oder Konzentrationen zu unempfindlich ist.
Für die Analytik von Pflanzenschutzmitteln kamen die Standardtechniken HPLC/DAD und GC/MS oder GC/µECD zum Einsatz. Des Weiteren wurde zur Anreicherung der Analyten die Festphasenextraktion (Solid Phase Extraction: SPE) verwendet. Für die Spuren- bzw. Rückstandsanalytik von Pestiziden mittels GC ist weiterhin in den meisten Fällen eine deutliche Anreicherung der Analyten notwendig, um eine hinreichende Detektionsempfindlichkeit zu erreichen. Die Anwendung eines großen Injektionsvolumens eröffnet hierbei gute Alternativen, sodass eine „large volume injection" in Form einer "programmed temperature vaporization (PTV)" verwendet werden sollte.
2.2 Material und Methoden
Zur Methodenentwicklung wurden die Festphasen Bond Elut® PPL 500 mg 3 ml sowie LiChrolut? EN 200 mg 3 ml (Varian, Darmstadt bzw. Merck, Darmstadt bezogen. Alle verwendeten Lösungsmittel und Chemikalien waren von einer zur Rückstandsanalyse geeigneten Reinheit (z. B. Lichrosolv® oder Suprasolv® , Merck, Darmstadt). Die Referenzsubstanzen (Analyten und die deuterierten internen Standards Metolachlord5 und Pendimethalin-d6) wurden von Sigma Aldrich (Steinheim) bzw. Ehrenstorfer (Augsburg) bezogen. Zur flüssigchromatographischen Analyse wurde ein HPLC Agilent 1100 System (Agilent, Waldbronn), bestehend aus einer binären Pumpe G1312A, Säulenofen G1316A mit Säulenschaltventil, thermostatisierbarem Probengeber G1329A mit G1330B sowie einem Diodenarray-Detektor DAD G1315B verwendet. Zur Reinigung des wässrigen Eluenten (Phosphatpuffer) diente ein herkömmliches Kieselgel C18 Material (Reinigungssäule).
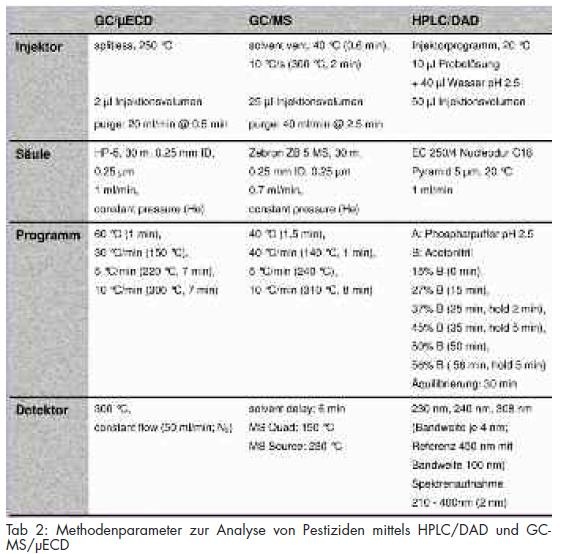
Die gaschromatographischen Untersuchungen erfolgten mit einem Agilent 6890N Network GC System mit µECD und 5973 Network Mass Selective Detector. Zur Probenaufgabe wurde ein Multi Purpose Sampler (MPS 2) der Fa. Gerstel (Mülheim a. d. Ruhr) genutzt. Die verwendeten Geräteeinstellungen und Parameter zur Analyse sind in Tab 2 aufgelistet.
2.3 Bestimmung polarer Herbizide mittels HPLC-DAD
Ein Liter Trinkwasserprobe wurde nach Einstellung des pH-Wertes auf 4,0 und Austreiben gelöster Luft im Ultraschallbad mit einer Flussrate von etwa 2 ml/min mittels Bond Elut? PPL 500 mg 3 ml extrahiert. Nach Beendigung der Extraktion wurden die Festphasensäulen mit 1 x 2 ml Wasser pH 4,0 gewaschen, im Stickstoffstrom für 15 Minuten getrocknet und mit 10 x 1 ml Methanol eluiert. Das Eluat wurde im Stickstoffstrom vorsichtig bis zur Trockne eingeengt und der Rückstand in 125 µl Acetonitril aufgenommen. Der so erhaltene Probenextrakt wurde mit den in Tab 2 angeführten Einstellungen mittels HPLC/DAD untersucht.
2.4 Bestimmung von Herbiziden und Fungiziden mittels PTV-GC-MS/µECD
Zu einem Liter Trinkwasserprobe wurden 50 ng der deuterierten internen Standardsubstanzen Metolachlor-d5 und Pendimethalin-d6 zugesetzt. Nach Einstellung des pH-Wertes auf pH 7, wurde die gelöste Luft im Ultraschallbad ausgetrieben. Mit einer Flussrate von 3 bis 4 ml/min wurde die Probe mittels der SPE (LiChrolut? EN 200 mg 3 ml) extrahiert. Nach Beendigung der Extraktion wurden die Festphasensäulen durch Anlegen von Vakuum für ca. 15 Minuten getrocknet. Die Elution erfolgte mit 5 ml Methanol und 25 ml Aceton. Das Eluat wurde nach Zugabe von 50 ng Aldrin als Retentionszeitmarker für die Analyse mittels GC/µECD am Rotationsverdampfer bei max. 40 °C und 300 mbar vorsichtig bis zur Trockene eingeengt und der Rückstand in 1 ml Hexan aufgenommen. Nach Trocknung mit Natriumsulfat wurden gleiche Volumina in GC-Vials mit Mikroinlet und Bördelkappe abgefüllt und mit den in Tab 2 dargestellten Parametern gaschromatographisch mittels massenselektiver Detektion und µECD untersucht.
3. Ergebnisse und Diskussion
3.1 Methodenentwicklung HPLC/DAD
Die zur HPLC-Bestimmung vorgesehenen Substanzen gehören der Gruppe der Harnstoffderivate an und sind in Tab 1 gekennzeichnet. Zusätzlich wurden die Substanzen Atrazin, die Metaboliten Desisopropyl- und Desethylatrazin sowie Diuron aufgrund ihrer Umweltpersistenz mit in die Methode aufgenommen. Bedingt durch die chemischen Eigenschaften der Substanzen, wie z. B. geringe UV-Absorption, Lage der Maxima in den UV-Spektren, und unter Berücksichtigung der Reinheit der verwendeten Reagenzien wurde die Umkehrphasenchromatographie (reversed phase; RP) an einer Nucleodur®-Säule gewählt. Als Fließmittel wurde ein Gradient aus Phosphat-Puffer (pH 2.5) und Acetonitril gewählt. Die Reinheit der Suprapur-Qualitäten des verwendeten Kaliumdihydrogenphosphates und der ortho-Phosphorsäure 85 % reichten für den Nachweis der Herbizide aufgrund von Stör-Peaks nicht aus. Durch Einbau einer Reinigungssäule in den Lösungsmittelkanal des Phosphatpuffers konnte dieses Problem jedoch behoben werden.
Im Zuge der Methodenentwicklung erwies sich eine C18-Phase mit polarem Endcapping gegenüber einer C8-Phase mit Endcapping und einer C18-Amide- embedded Phase mit fused core- Technologie als überlegen. Eine HILICPhase (hydrophilic interaction liquid chromatography) im Zwitterion-Zustand brachte keine zufriedenstellende Trennung. Die Polarität der Analyten war für diese Art der Chromatographie nicht ausreichend, sodass eine sehr frühe Elution und schlechte Auflösung resultierten. Auch eine Änderung des pHWertes und Optimierung der Pufferkonzentration führten nicht zum gewünschten Erfolg. Nach einer Temperaturerhöhung der RP-Säulen auf 35 °C wurde ein Abbau einzelner Substanzen während der Chromatographie festgestellt und die Ergebnisse waren somit nicht reproduzierbar.
Die Standardlösungen von Nicosulfuron, Thifensulfuron-methyl, Rimsulfuron und Amidosulfuron in Millipore? Reinstwasser veränderten sich im HPLC-Vial innerhalb von 48 Stunden. In Phosphatpuffer- Lösung pH 2,5 war Rimsulfuron innerhalb von 14 Stunden abgebaut, Flazasulfuron war nur noch zu 50 % vorhanden. Daher wurden zur Vermeidung der geschilderten Degradation alle Messlösungen in Acetonitril angesetzt und die Injektion erfolgte mit einem Injektorprogramm, mit dem die Messlösungen mit Wasser pH 2,5 in der Aufgabekapillare gemischt und anschließend injiziert wurden. Auf die Mischung mit Phosphatpuffer-Lösung wurde verzichtet, um ein Auskristallisieren von Phosphaten bei hohem organischem Anteil und Verstopfen der Aufgabekapillare zu verhindern.
Zur Anreicherung der Analyten aus Trinkwasser wurden SPE-Phasen auf der Basis von Silica non endcapped C18, Polystyrene-divinylbenzene (PS/DVB), PS/DVB+WAX, N-Vinylpyrrolidon/ DVB, N-Ethylpiperidon/PS/ DVB und functionalized PS/DVB getestet. Zur Auswahl einer geeigneten Festphase wurden neben der Adsorption der Substanzen (Wiederfindungsrate) auch mitextrahierte Störsubstanzen aus den verwendeten Materialien betrachtet. Die Festphase Bond Elut® PPL (functionalized PS/DVB) wies die geringsten Störeinflüsse sowie die besten Wiederfindungsraten auf. Da die Harnstoffherbizide überwiegend schwach saure funktionelle Gruppen besitzen, war für eine reproduzierbare und gute Wiederfindungsrate der pH-Wert bei den Trinkwasserproben zwingend auf pH 4,0 einzustellen. Um eine kontinuierliche Benetzung der Festphase zu gewährleisten, mussten gelöste Luft und CO2 durch Einwirkung von Ultraschall anschließend entfernt werden. Weiterhin wurde beobachtet, dass ein zu langes Trocknen der Festphase nach der Extraktion der Wasserproben die Wiederfindung einiger Substanzen reduzieren kann. Mit dem hier vorgestellten optimierten Verfahren wurden reproduzierbare, präzise Ergebnisse erhalten, die die genannten Vorgehensweisen und Bedingungen berücksichtigen.
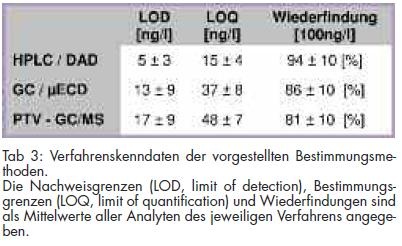
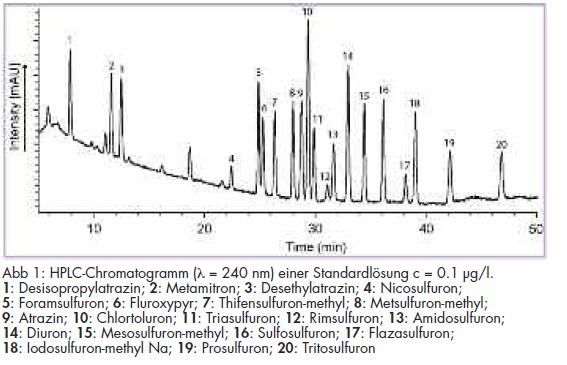
Zur Methodenvalidierung wurden neben der Bestimmung der Linearität der Bestimmungsmethode Wiederfindungsexperimente mit dotierten Wasserproben bei 100 ng/l und 30 ng/l durchgeführt. Die Identifizierung der Analyten erfolgte über die Retentionszeiten. Durch die zeitgleiche Aufnahme von UV-Spektren ließen sich neben der Retentionszeit hierüber die Substanzen identifizieren. Die Messung der Analyten bei drei UV-Wellenlängen ergab sich aus deren Absorptionsmaxima. Der Gehalt wurde aus einer externen Kalibrierreihe von 25 - 200 ng/l aus den Flächen der Analyten mit linearer Regression ermittelt. Aus den erhaltenen Daten wurden Präzision, Wiederfindungsraten und Nachweisgrenzen der Analysenmethode bestimmt. Die resultierenden Verfahrenskenndaten sind in Tab 3 dargestellt. Ein Beispielchromatogramm eines Kalibrierstandards ist in Abb 1 wiedergegeben.
3.2 Methodenentwicklung PTV-GC/MS und GC/µECD
Die zur gaschromatographischen Bestimmung geeigneten Substanzen wurden nach ihren physikalisch-chemischen Eigenschaften (saure oder basische funktionelle Gruppen, Chromatographierbarkeit mit oder ohne Notwendigkeit zur Derivatisierung) unterteilt. Sie wurden weiterhin aufgrund ihres Anteils an Halogenatomen und damit der Möglichkeit zur Detektion mittels µECD charakterisiert.
Die wesentliche Unterteilung erfolgte nach basischen bzw. überwiegend neutralen sowie sauren Substanzen. Für den gaschromatographischen Nachweis sind saure funktionelle Gruppen in jedem Fall zu derivatisieren und für die Probenextraktion mittels SPE muss der pH-Wert der Probe eingestellt werden. Da es sich bei dem Großteil der ausgewählten Verbindungen um neutrale Substanzen handelte, wurden diese zunächst zur Methodenentwicklung ausgewählt. Für saure Stoffe sollte anschließend eine eigene Methode entwickelt werden, da aufgrund der physikalisch-chemischen Eigenschaften eine eigenständige Probenaufarbeitung erfolgen muss.
In Tab 1 sind die mittels Gaschromatographie bestimmten Substanzen gekennzeichnet. Im Verlauf der chromatographischen Methodenentwicklung stellten sich einige Substanzen, z. B. Mesotrion und Sulcotrion, durch starke aber unspezifische Fragmentierungen und die hieraus resultierende geringe Detektionsempfindlichkeit als ungeeignet zur massenselektiven Detektion heraus. Ebenso zeigten verschiedene Fungizide trotz vorhandener Halogenatome nur geringe Detektionsempfindlichkeiten am µECD. Die Fungizide Captan und Folpet seien des Weiteren an dieser Stelle für sehr hydrolytisch empfindliche Substanzen angeführt, die innerhalb weniger Stunden in Wasser abgebaut werden. Daher war es wenig sinnvoll, diese Analyten in die Methode einzubinden.
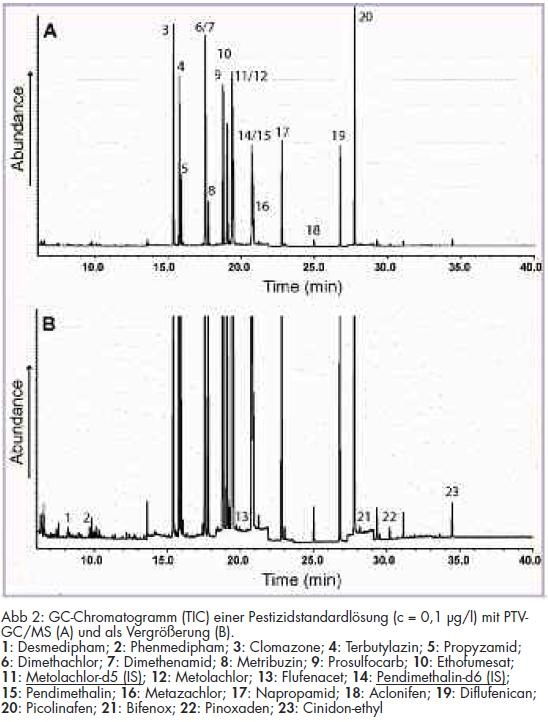
An diesem Beispiel wird deutlich, wie wichtig die Kenntnis ökochemischer Vorgänge, wie Metabolisierung und Abbau von Pestiziden, ist. Insbesondere bei der massenselektiven Detektion zeigte sich, dass zum Erreichen der Nachweisgrenze von < 25 ng/l gemäß TrinkwV 2001 die üblicherweise genutzte Injektionsmenge von 1 bzw. maximal 2 µl nicht ausreicht. Auch ein stärkeres Einengen des nach SPE erhaltenen Extrakts trug nur unwesentlich zu einer Verbesserung bei, zumal das Extraktvolumen groß genug sein muss, da zwei instrumentelle Verfahren mit entsprechenden Spülschritten der Injektionsnadel angewendet werden mussten. Aus diesem Grund wurde die Injektion größerer Injektionsvolumina angestrebt. Hierzu konnte die PTV für eine große Volumenaufgabe erfolgreich angewendet und das Injektionsvolumen um das 25-fache gesteigert werden (vgl. Abb 2).
Zur Probenextraktion mittels SPE wurden verschiedene Festphasen untersucht. Hierzu gehörten sowohl C18- Phasen auf Kieselgel- und Polymerbasis (z. B. Chromabond® HRX, C18 Hydra, Easy oder LiChrolut® RP-18, EN) als auch Carbonphasen (z. B. Chromabond® Carbon oder ENVI™ Carb) mit Backflush-Elution.
Die besten Resultate zur Extraktion der Analyten für beide apparative Bestimmungen, der GC/MS und der GC/µECD, wurden dabei mit LiChrolut® EN 200 mg 3 ml erhalten. Interferenzen, die bei Verwendung anderer Festphasen insbesondere bei der massenselektiven Detektion auftraten, konnten vermieden werden. Die Anwendung von Carbonphasen zeigte gute Extraktionsausbeuten, jedoch waren zur Desorption einerseits eine Backflush- Elution und andererseits große Lösungsmittelvolumina notwendig. Dies führte zu einer sehr aufwändigen Probenvorbereitung, da jede Festphase einzeln eluiert werden musste. Weiterhin ist aus ökonomischen Gesichtspunkten die Verwendung großer Lösungsmittelvolumina nicht sinnvoll.
Die Extraktion der Analyten aus 1 Liter Wasser konnte unter Verwendung von LiChrolut® EN nach einem Konditionierungsschritt mit 3 Säulenvolumina Aceton, 3 Säulenvolumina Methanol und 2 Säulenvolumina bidestillierten Wassers innerhalb weniger Stunden erfolgen. Die Analyten wurden mit 5 ml Methanol und 25 ml Aceton eluiert und das Eluat am Rotationsverdampfer schonend eingeengt. Die Analyten wurden in Hexan als leicht flüchtigem und für die GC gut geeignetem Lösungsmittel aufgenommen. Verbliebenes Restwasser aus der SPE konnte mit Natriumsulfat erfolgreich und einfach entfernt werden. Alternativ wurde 2,2-Dimethoxypropan als Wasser bindendes Reagenz getestet, welches aber keine Vorteile erbrachte. Die Verwendung von Methanol als Lösungsmittel führte bei einigen Substanzen nicht zu reproduzierbaren Ergebnissen infolge einer wahrscheinlich alkoholytischen Zersetzung im Injektor.
Zur Korrektur von Analytverlusten während der Probenvorbereitung wurden für die GC/MS zwei deuterierte interne Standardsubstanzen, Metolachlord5 und Pendimethalin-d6, verwendet. Die Auswertung bezüglich Identifizierung und Konzentration der Analyten erfolgte über die relative Retentionszeit sowie den Flächenquotienten aus Analyt und internem Standard mittels linearer Regression von Kalibrierstandards im Konzentrationsbereich von 25 - 200 ng/l. Zum Nachweis von Pestiziden mittels GC/µECD wurde Aldrin als Retentionszeitmarker nach der Elution der Festphasensäulen zugesetzt, um die relative Retentionszeit zu bestimmen, da keine geeignete Substanz als interner Standard ermittelt werden konnte. Die Kalibrierung erfolgte im oben genannten Konzentrationsbereich ohne Korrektur durch einen internen Standard ebenfalls mit linearer Regression der Flächen der Analyten zur Konzentration.
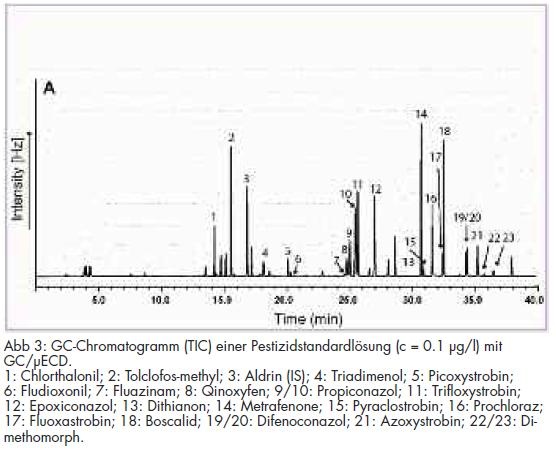
Zur Methodenvalidierung wurden neben der Bestimmung der Linearität der Bestimmungsmethode Wiederfindungsexperimente bei 100 ng/l und 50 ng/l durchgeführt. Aus den ermittelten Daten wurden die Präzisionen, die Wiederfindungsraten und die Nachweisgrenzen der Analysenmethode ermittelt. Die resultierenden Ergebnisse sind in Tab 3 dargestellt. Beispielchromatogramme von Kalibrierstandards für die Bestimmung mit GC/MS und GC/µECD sind in den Abb 2 und 3 wiedergegeben.
4. Schlussfolgerungen
Für den Nachweis von Pestiziden in Trinkwasser konnten verschiedene chromatographische Strategien entwickelt werden. Durch kombinierte Analyse gelingt es derzeit, insgesamt 54 von 111 relevanten Pestiziden mittels HPLC/DAD und GC/MS bzw. GC/µECD nachzuweisen. Für zwei weitere Herbizide, Glyphosat, seinem Metaboliten AMPA und Glufosinat, wurde vor einiger Zeit eine eigenständige Methode entwickelt, die aufgrund der sehr polaren Eigenschaften der Analyten und der damit verbundenen speziellen Probenaufarbeitung notwendig war [6].
Der Nachweis von Rückständen im Trinkwasser sowie die Festlegung eines definierten Substanzspektrums stellt aufgrund der teilweise geringen Stabilität und des raschen Abbaus von Pestiziden im Trinkwasser nach wie vor eine Herausforderung dar. Wie am Beispiel der Phthalimid-Fungizide Captan und Folpet gezeigt wurde, müssen die Pestizide auf ihre ökochemischen Eigenschaften geprüft werden, um nicht fälschlicherweise Substanzen zu analysieren, die im Trinkwasser nur als Metaboliten vorliegen können. Daher sind in Anlehnung an die Richtlinie 91/414/EWG relevante Metaboliten in die Analyse einzubeziehen [7].
Diese Betrachtung des Pestizidspektrums und die Erweiterung des Analysenumfangs sind daher für die effiziente und ökonomische Analyse wichtig und zu berücksichtigen. Hierbei wird das Stoffspektrum auf Herbizide mit sauren funktionellen Gruppen, wie die Carbonsäurederivate (z. B. MCPA), ausgeweitet, die einen wesentlichen Anteil einnehmen. Eine Methode zur Bestimmung von sauren Herbiziden, so genannten Phenoxyalkancarbonsäuren, wurde vor etwa 2 Jahren am ZInst- SanBw KOB entwickelt [8]. Neben den bereits mit dieser Methode analysierbaren Analyten Bentazon, Dichlorprop- P, MCPA, Mecoprop-P, Bromoxynil, Dicamba, Ioxynil und Clopyralid sollen neuere Substanzen in diese Methode integriert werden, wobei die Grundzüge der Probenvorbereitung und der analytische Nachweis auf der vorhandenen Methode aufbauen sollen.
Das hier dargestellte Konzept hat gezeigt, dass die Analyse von Pestiziden in Trinkwasser aufwändig ist und stetiger Optimierungen und Anpassungen der Verfahrensabläufe aufgrund neuer und auslaufender Zulassungen der Pestizide sowie deren Abbauprodukte bedarf. Zukünftig werden daher geeignete Screeningverfahren zur schnellen Detektion von relevanten Analyten eine entscheidende Rolle einnehmen, da nur hiermit Analysenziel und Arbeitsaufwand vereinbar sind.
Durch die gezielte Kombination der Probenvorbereitung mit sensitiven chromatographischen Methoden wird es gelingen, das Untersuchungsspektrum der Pestizide stetig zu erweitern und einen Beitrag zum vorbeugenden Gesundheitsschutz im Grundbetrieb und weltweiten Einsatz der Bundeswehr zu leisten.
Literatur:
- Trinkwasserverordnung 2001, BGBl. I S. 2407 (TrinkwV 2001)
- Grundwasserdatenbank Wasserversorgung: Verlagerungs- und Abbauverhalten ausgewählter Sulfonylharnstoff- Herbizide im Boden http://www.grundwasserdatenbank. de/bilder/pdf/Verlagerungs- und Abbauverhalten ausgewählter Sulfonylharnstoff- Herbizide im Boden.pdf
- Bundesamt für Verbraucherschutz und Lebensmittelsicherheit - Absatz an Pflanzenschutzmitteln in der Bundesrepublik Deutschland. Ergebnisse der Meldungen gemäß § 19 Pflanzenschutzgesetz für das Jahr 2008
- Pflanzenschutzgesetz in der Fassung der Bekanntmachung vom 14. Mai 1998 (BGBl. I S. 971, 1527, 3512), zuletzt geändert durch Artikel 13 des Gesetzes vom 29. Juli 2009 (BGBl. I S. 2542)
- FOOTPRINT: creating tools for pesticide risk assessment and management in Europe. http://sitem.herts.ac.uk/aeru/footprint/en /index.htm
- Küsters M, Gerhartz M: J Sep Sci 2010; 33: 1139-1146
- Richtlinie 91/414/EWG des Rates vom 15. Juli 1991 über das Inverkehrbringen von Pflanzenschutzmitteln. Amtsblatt Nr. L 230 vom 19/08/1991 S. 0001 - 0032
- Bimber U, Küsters, M, Klünder R, Gerhartz M: Öffentlich - rechtliche Überwachung in der Wehrpharmazie im Jahr 2007. München: Sanitätsamt der Bundeswehr, Abteilung VIII Wehrpharmazie 2008; 73-78
Datum: 01.10.2010
Quelle: Wehrmedizinische Monatsschrift 2010/10