ENTSCHLÜSSELUNG HOCHPATOGENER MIKROBEN: VOLLGENOMSEQUENZIERUNG IN DER BUNDESWEHR
Deciphering highly pathogenic microbes: Whole-Genome Sequencing in the Bundeswehr
Aus dem Institut für Mikrobiologie der Bundeswehr, München (Leiter: Oberstarzt Prof. Dr. L. Zöller)
Markus Antwerpen und Enrico Georgi
WMM, 58. Jahrgang (Ausgabe 2/2014; S. 34-38)
Zusammenfassung
Die Vollgenomsequenzierung von Mikroorganismen in Verbindung mit Bioinformatik ist eine Schlüsseltechnologie für die Aufklärung ungewöhnlicher Krankheitsausbrüche. Dieser Beitrag stellt die Methode im Kontext moderner klinischer Mikrobiologie vor und zeigt Anwendungsbeispiele, wie sie am Institut für Mikrobiologie der Bundeswehr, in dessen Verantwortungsbereich die Aufklärung solcher Ereignisse liegt, durch den neu aufgestellten Funktionsbereich „Mikrobielle Genomik und Bioinformatik“ bearbeitet werden.
Das Ziel liegt dabei nicht nur auf der Erstellung gerichtsverwertbarer genetischer Fingerabdrücke von Bakterien zur Rückverfolgungsanalyse, sondern auch im Einsatz neuartiger Untersuchungsverfahren, die die Diagnostik erkrankter Patienten unterstützen.
Schlüsselwörter: Bioforensik, Genomik, Bioinformatik, Mikrobiologie, Sequenzierung
Summary
Whole-Genome-Sequencing of microorganisms in combination with bioinformatics has become a key technology for reconnaissance of unusual disease outbreaks. This study puts this new method into the context of modern clinical microbiology. We present examples of application, conducted by the new division “Microbial Genomics and Bioinformatics” at the Bundeswehr Institute of Microbiology. The division’s mission is not only to generate court-proof genetic fingerprints of microbes for trace-back-analysis, but also the development of new approaches for supporting disease diagnostics.
Keywords: Bioforensics, Genomics, Bioinformatics, Microbiology, Sequencing
Fallberichte
06.12.2009, Aachen, Deutschland: Ein 42-jähriger Mann wird mit schmerzhafter Schwellung der unteren Extremität nach intravenöser Heroinapplikation initial mit Verdacht auf eine tiefe Beinvenenthrombose stationär aufgenommen. Neben chronischem Abusus von i.v.-Drogen, Alkohol und Benzodiazepinen sind eine chronische Hepatitis B und C sowie eine HIV-Infektion bekannt. Computertomographische Aufnahmen zeigen eine Muskelnekrose mit drohendem Kompartmentsyndrom. Trotz chirurgischer Intervention bei akuter nekrotisierender Fasciitis und antibiotischer Behandlung mit Meropenem erliegt der Mann am 13. Dezember einem multiplen Organversagen [1]. Erst am 18. Dezember werden aus einem Wundabstrich gram-positive Endosporen-bildende Bakterien als Bacillus anthracis, einem als Biowaffen-Agens der Kategorie A gelisteten Erreger, bestätigt. Die infektionsepidemiologischen Kompetenzzentren Robert-Koch-Institut (Berlin) und Friedrich-Löffler-Institut (Jena) sowie der gesamte öffentliche Gesundheitsdienst stehen damit vor dem ersten humanen Milzbrandfall in Deutschland seit 1994.
Fast zeitgleich verstirbt am 16.12.2009 ein Mann nach Heroininjektion und nachfolgender Milzbrandinfektion in Glasgow, Schottland; ein weiterer wird dort ins Krankenhaus eingeliefert. Die britischen Behörden geben daraufhin im Early Warning und Reporting System (EWRS) der europäischen Seuchenschutzbehörde ECDC eine Warnmeldung zu einem Milzbrandausbruch unter i.v.-Drogenabhängigen heraus [2]. Forscher aus Europa und den USA arbeiten mit den Nachrichtendiensten zusammen, um Indizien über die Herkunft des Heroins und der Milzbrandstämme zu erhalten. Auch das Institut für Mikrobiologie der Bundeswehr ist an den Untersuchungen beteiligt. Epidemiologen in Europa erwarten das Auftreten weiterer Fälle und sollen Recht behalten. Die traurige Bilanz Ende 2011: 6 Tote in England, 13 in Schottland, 2 in Deutschland.
05.06.2012, Regensburg, Deutschland: Ein drogenabhängiger Mann verstirbt in Regensburg kurz nach Aufnahme in ein Krankenhaus. Aus der Blutkultur werden Bakterien isoliert und vorläufig als Bacillus anthracis identifiziert. Eine Probe wird an den Zentralbereich Diagnostik des Instituts für Mikrobiologie der Bundeswehr in München gesendet. Unmittelbar nach Bestätigung der Diagnose Milzbrand werden Typisierungsuntersuchungen veranlasst, um den Stamm eingehend zu charakterisieren und mit bestehenden Datenbankeinträgen zu vergleichen [3]. Zusätzlich zu den festgelegten Meldewegen wird die wissenschaftliche Community via www.ProMEDmail.org informiert. Auch Frankreich, Dänemark, England, Wales und Schottland melden im Verlauf 2012/2013 elf weitere Milzbrandfälle unter sogenannten PWIDs (people who inject drugs).
Und wieder sind es offene Fragen, die zunächst unbeantwortet im Raum stehen: Wo kommt das Heroin her? Ist es ein bewusstes Einbringen von Milzbranderregern in den Kreis von Heroinkonsumenten oder nur eine unbeabsichtigte Kontamination? Handelt es sich um den gleichen Stamm wie 2009/2010? Neue bislang für Europäer eher abstrakte Fragestellungen werden Realität und müssen beantwortet werden. Dafür muss auf neueste labortechnische Verfahren zurückgegriffen werden. Der Focus liegt dabei auf dem Identifizieren neuer für den Ausbruchsstamm einzigartiger selbst kleinster genetischer Unterschiede, die durch Fehler der bakteriellen DNA-Polymerase entstehen. Die Unterschiede können sich sowohl natürlich beispielsweise während des Lebenskreislaufes in der Natur etabliert haben oder es kann sich auch um fremd induzierte Mutationen handeln, wie sie bei Laborstämmen häufiger anzufinden sind. Im Vergleich mit Datenbanken sollen diese Unterschiede Hinweise geben, aus welcher Region das Bakterium stammt oder ob es sich gar um einen Laborstamm handelt. Am Institut für Mikrobiologie der Bundeswehr liegen – je nach Untersuchungsverfahren – genetische Fingerabdrücke für einen solchen Abgleich von bis zu 4000 Bacillus-anthracis-Isolaten aus der ganzen Welt vor. Die Schlüsseltechnologie hierfür bildet die Vollgenomsequenzierung von hochpathogenen Erregern in Verbindung mit Bioinformatik.
Einsatz molekularer Mikrobiologie für den Patienten
Zur Erregeridentifizierung setzt die klinische Mikrobiologie bis dato als Goldstandard auf Methoden der kulturellen Anzucht und biochemischen Differenzierung. Auch wenn in den letzten Jahren neue technische Verfahren wie beispielsweise MALDI-TOF (Matrix-Assisted Laser Desorbtion/Ionization - Time Of Flight) Massenspektrometrie die diagnostischen Algorithmen beschleunigt haben und optimierte Arbeitsabläufe (u.a. elektronische Befundübermittlung) Standard geworden sind, vergehen zwischen Probennahme am Patientenbett und endgültigem Befund mit Antibiogramm meist zwei Tage.
Insbesondere bei schwerkranken Patienten werden ergänzend Strategien der molekularen Identifizierung eingesetzt, um schneller ein Resultat zu erhalten. Dabei werden zunächst – sofern in der Probe vorhanden – hochspezifische kurze Nukleinsäure-Sequenzen der vermuteten Krankheitserreger exponentiell vermehrt (PCR) und anschließend mit verschiedenen Methoden nachgewiesen. Da dieses sehr sensitive Verfahren aber nur assay-spezifische Gensignaturen identifiziert, ist es zumeist auf häufige und/oder hochpathogene Erreger bzw. Zielgene beschränkt. Demgegenüber bietet die Vollgenomsequenzierung, also die ungezielte Anreicherung und Sequenzierung der (nahezu) kompletten genetischen Information eines Erregers, den Vorteil, selbst bei veränderten Sequenzmotiven oder neu auftretenden Pathogenen eine Identifizierung zu ermöglichen. Eine Auswahl der auszuwertenden Genabschnitte erfolgt – sofern gewünscht – erst bei der Auswertung der Rohdaten im Sinne einer Datenfilterung. Prinzipiell liegt aber die gesamte Erbinformation vor, die zur weiteren Charakterisierung herangezogen und entschlüsselt wird. Aus diesen Nebenbefunden können dann wiederum PCR-Assays abgeleitet werden, um gleichartige Erreger hochspezifisch und sensitiv nachweisen zu können.
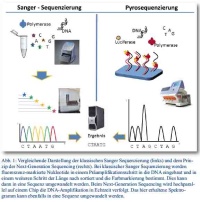
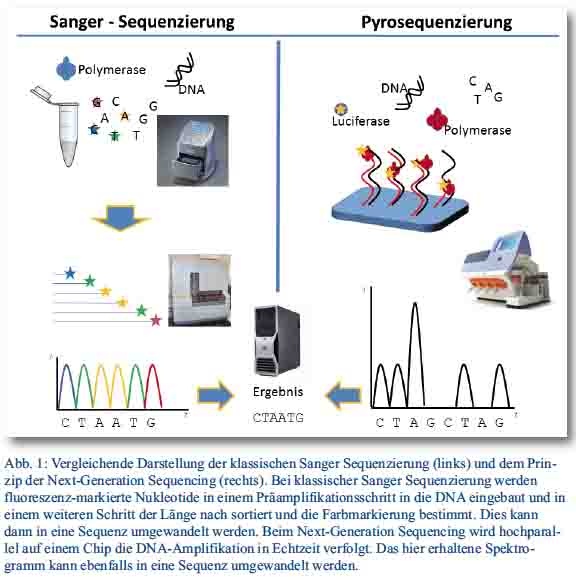
Entwicklung der Sequenziertechnologien
Die ersten für größere Projekte geeigneten Methoden zur DNA-Sequenzierung wurden bereits 1977 von Sanger et al. (Prinzip der Kettenabbruchsynthese) bzw. Maxam und Gilbert (Prinzip der basenselektiven chemischen Degradation) veröffentlicht. Durch höhere Genauigkeit und schnellere Resultate konnte sich das Sanger-Prinzip (Abbildung 1, links) durchsetzen und dominierte insbesondere nach Einführung von Fluoreszenz-markierten Dideoxynukleotiden (ddNTPs), Umstellung von Gel- auf Kapillarelektrophorese und automatischer Auswertung jahrzehntelang das Feld der Sequenzierung. Die Ergebnisse nach dieser Methode zeigen eine hohe Qualität; jedoch ist diese Art der Technologie, mit der u. a. das erste menschliche Genom sequenziert wurde, für Vollgenomanalysen sehr kosten- und äußerst zeitintensiv.
Deutlich schneller werden heutzutage Sequenzen generiert, indem Millionen kurzer DNA-Fragmente parallel sequenziert werden. Um die neuen Methoden von der klassischen Sequenzierung abzugrenzen, wurde der Begriff Next-generation sequencing (NGS) eingeführt. Die zugrundeliegende Methode der Pyrophosphat-Detektion wurde bereits 1985 beschrieben und so weiterentwickelt, dass die von Jonathan Rothberg gegründete Firma „454 Life Science“ die erste kommerzielle Next-generation Sequenzierplattform im Jahr 2005 auf den Markt bringen konnte. Die sogenannte Pyrosequenzierung nutzt die Abspaltung eines Pyro-Phosphat-Ions (PPi) beim Anhängen eines Nukleotids an den zu synthetisierenden DNA-Strang. Dieses Ion kann nun – je nach verwendetem System – beispielsweise photometrisch unter Nutzung von Luciferase detektiert werden (Abbildung 1, rechts). Zur Verstärkung des Signals geht der eigentlichen Sequenzierreaktion eine Amplifikation der einzelnen DNA-Fragmente voraus (emulsion PCR).
Das am Institut für Mikrobiologie der Bundeswehr eingesetzte System Ion Torrent Personal Genome Machine nutzt die Halbleitersequenziertechnologie. Anstelle von Lichtsignalen werden über Feldeffekttransistoren während der Sequenzierreaktion geringste pH-Veränderungen detektiert [4], wodurch die Rohdaten schneller als bei anderen Plattformen gewonnen werden können. Das System kann seine Stärken deshalb insbesondere bei der Ausbruchsaufklärung ausspielen. Bereits 2011 konnte das Team um Prof. Harmsen damit im Rahmen der EHEC/HUS-Epidemie in Deutschland den Ausbruchsstamm in nur 62 Stunden vollständig sequenzieren [5]. Die kontinuierliche Weiterentwicklung der verwendeten Chips samt passenden chemischen Reagenzien verbessert die Resultate der Sequenzierung insbesondere im Hinblick auf Leselänge und Parallelität stetig.
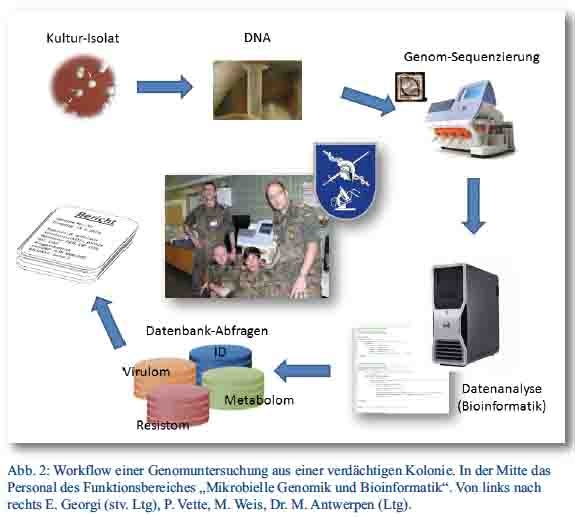
Erst mit Einführung der kleineren und kostengünstigeren Benchtop-Geräte traten die NGS-Plattformen ihren Siegeszug an und haben inzwischen in vielen Laboren Einzug gehalten. Die Sequenzierung mikrobieller Genome ist heutzutage schnell und vergleichsweise kostengünstig geworden. Im Rahmen eines Sonderforschungsprojektes war es 2013 möglich, ein Vollgenom-Sequenziergerät für das Institut zu beschaffen, Personal in der Anwendung zu schulen und Arbeitsabläufe aufzustellen (Abbildung 2).
Schwerpunkte der Projekte für die Bundeswehr
Als Ressortforschungseinrichtung im Geschäftsbereich des Bundesministers der Verteidigung liegt der wissenschaftliche Fokus am Institut für Mikrobiologie der Bundeswehr auf anwendungsorientierter Forschung auf dem Gebiet des medizinischen B-Schutzes. Wichtige Forschungsfelder sind die schnelle und zweifelsfreie Diagnose von Gesundheitsstörungen durch biologische Kampfstoffe oder vergleichbare Noxen, die wissenschaftliche Aufklärung unklarer Ausbruchsgeschehen, die forensische Verifikation möglicher B-Kampfstoff-Freisetzungen sowie die Implementierung wirksamer Kontrollmaßnahmen in biologischen Gefährdungslagen. Mit der Einrichtung eines Zentralbereichs Diagnostik, der Implementierung eines QM-Systems und Akkreditierung der diagnostischen Leistungen nach DIN ISO 15189, der Weiterentwicklung der modularen, weltweit schnellverlegbaren Labor- und B-Aufklärungskapazitäten und dem Aufbau molekularer Typisierungsdatenbanken sind auf allen Gebieten große Fortschritte erzielt worden. Mit der Aufstellung eines neuen Funktionsbereichs „Mikrobielle Genomik und Bioinformatik“ zum 01.07.2013 werden nun die vielversprechenden neuen Sequenziertechnologien auf die Anwendbarkeit für wehrwissenschaftliche Fragestellungen untersucht und in das Portfolio des Leistungsspektrums am Institut integriert. Die am Institut für Mikrobiologie der Bundeswehr im Bereich Vollgenomsequenzierung laufenden und geplanten Projekte konzentrieren sich auf drei Bereiche:
(1) Anwendbarkeit der neuen Sequenziertechnologien im
klinisch-infektiologischen Kontext.
(2) Bioforensische Fragestellungen bei der Untersuchung
ungewöhnlicher Krankheitsausbrüche.
(3) Wissenschaftlich-wehrmedizinische Grundlagenforschung.
Klinisch-infektiologische Projekte
Im klinischen Kontext kann die Vollgenomsequenzierung vor allem dort ihre Stärken ausspielen, wo eine umfassende und schnelle Charakterisierung eines Krankheitserregers wichtig ist. Während in den meisten Fällen eine Identifizierung bis auf Speziesebene ausreicht, um gemeinsam mit den klinischen Kollegen eine optimale Therapiestrategie zu finden, reicht diese Diagnostiktiefe zur Aufklärung von Infektionsketten oft nicht aus. Daher überrascht es nicht, dass in den letzten Jahren gerade im Bereich Krankenhaushygiene einige Arbeiten publiziert wurden, die die neuen DNA-Sequenziertechnologien nutzen. Ein gutes Beispiel ist ein DNA-sequenzbasiertes Frühwarnsystem für den Nachweis von MRSA-Ausbrüchen auf Krankenhausebene, welches durch die Arbeitsgruppe um Prof. Harmsen am Universitätsklinikum Münster entwickelt wurde. Durch Kombination von schnell verfügbaren Typisierungsergebnissen mit spezieller bioinformatischer Software ist dabei eine automatisierte Differenzierung zwischen zufälligen Häufungen von MRSA-Fällen und Ausbrüchen möglich [6]. Verschiedene Studien zeigen, dass insbesondere Benchtop-Vollgenomsequenzierer zur Ausbruchsaufklärung und Surveillance von nosokomialen Erregern wie MRSA oder Clostridium difficile gut geeignet sind. Durch das höhere Auflösungsvermögen im Vergleich zu anderen Methoden können Übertragungswege viel besser nachvollzogen und dadurch spezifischere Maßnahmen zur Infektionskontrolle implementiert werden, um die weitere Verbreitung der Keime einzudämmen.
Infektionen mit dem „gleichen“ Erreger können höchst unterschiedlich verlaufen. Neben Übertragungsweg, Infektionsdosis und Wirtsfaktoren können spezifische Virulenzfaktoren diese verschiedenen klinischen Bilder erklären. Durch Sequenzierung und Entschlüsselung der genetischen Information lassen sich scheinbar gleiche Erreger, d.h. solche mit gleichem Identifizierungsergebnis auf Spezies- oder Subspezieslevel, nach ihren Virulenz- und Resistenzgenen differenzieren. Damit können die individuelle Prognose eines Patienten besser eingeschätzt sowie Therapienotwendigkeit und –optionen besser abgewogen werden.
Nicht zuletzt kommt es immer wieder zum Auftreten von neuen oder veränderten Infektionserregern bei Mensch und Tier, die sich aufgrund spezieller Kulturanforderungen oder veränderter Molekularstruktur den etablierten Nachweisverfahren entziehen. Mit der Next-Generation Sequencing-Technologie konnte 2011 ein Krankheitsausbruch in Milchviehbeständen Nordrhein-Westfalens mit einem bis dahin unbekannten Virus der Familie Bunyaviridae aufgeklärt werden. Aus den Vollgenomdaten konnten molekulare Tests abgeleitet und so weitere Infektionen mit dem als Schmallenberg-Virus bezeichneten Erreger aufgedeckt werden [7].
Bioforensische Projekte
Ein gutes Beispiel für einen ungewöhnlichen Krankheitsausbruch, bei dem auch die bioforensische Fragestellung von enormer Bedeutung war, ist die EHEC-Epidemie 2011 in Deutschland. Ungewöhnlich war dieser vor allem deshalb, weil der ursächliche Krankheitserreger erhebliche genetische Unterschiede zu denjenigen E.coli-Stämmen aufwies, die typischerweise mit Hämorrhagisch-urämischem Syndrom (HUS) assoziiert sind. Die Veränderungen im Genom führten zu einem neuen Phänotyp mit einer seltenen Kombination spezifischer Virulenzeigenschaften und speziellen Resistenzmustern, der in Verbindung mit der Verbreitung über Sprossenkeimlinge etwa 3000 Krankheitsfälle mit akuter Gastroenteritis und über 850 Fälle von HUS verursachte [8]. Wie lassen sich solche Veränderungen von Erregern und deren Ausbreitung erklären?
Auch das Eingangsszenario zeigt eindrücklich, dass immer wieder ungewöhnliche Cluster von Infektionen auftreten, bei denen eine nicht-natürliche Ursache diskutiert werden kann. Um solche bioforensische Fragestellungen in Zukunft noch besser beantworten zu können, hat sich das Institut für Mikrobiologie der Bundeswehr in den letzten Jahren intensiv mit deutschen, europäischen und internationalen Partnern vernetzt. So ist das Institut beispielsweise Teil des European Biodefence Laboratory Network (EBLN) der europäischen Verteidigungsministerien, in dem Methoden zu Charakterisierung, Vergleich und Rückverfolgung europäischer Pathogene harmonisiert und weiterentwickelt werden. Eine besondere Bedeutung kommt dabei der Implementierung eines Qualitätsmanagements für wissensbasierte Dienstleistungen zu, d. h. der Festlegung von Methoden zur standardisierten Probenbearbeitung und anschließender Analyse vollgenomischer Informationen.
Bei dieser Computer-gestützen Analyse liegt der Augenmerk auf genetischen Stamm- oder Isolat-spezifischen Merkmalen. Neben Punktmutationen (SNPs, Single Nucleotide Polymorphisms) wird auch der genetische Fingerabdruck des Isolates bestimmt. Dabei kommt die Methode MLVA (Multiple Locus Variable Number of tandem repeat Analysis) zur Anwendung, wie auch bei Vaterschaftsanalysen in der Humangenetik.
Die in der Mitte des 19. Jahrhunderts begründeten klassischen Methoden der Infektionsepidemiologie wie z. B. geographisches Mapping sind im 20. Jahrhundert verfeinert worden. Erst durch die Fortschritte in der Molekularbiologie in den letzten Jahrzehnten hat sich ein neues Feld etabliert: Die molekulare Epidemiologie kann durch hochauflösende Charakterisierung pathogener Organismen zur Abschätzung der Diversität der Erreger und damit zur Ausbruchsaufklärung beitragen. Durch die technische Revolution der Sequenziertechniken können innerhalb immer kürzerer Zeit immer größere Datenmengen zu Untersuchungsproben gewonnen werden. Die Auswertung dieser Datenflut stellt aktuell eine der größten Herausforderungen der modernen Mikrobiologie dar und ist nur mit bioinformatischen Methoden zufriedenstellend lösbar.
Wehrwissenschaftliche Projekte
Aufgrund der technischen Limitationen etablierter molekularbiologischer Methoden war die bisherige Mikrobiologie bei genetischen Analysen auf einzelne kultivierbare Erreger konzentriert, und dabei v. a. auf solche mit human- oder veterinärmedizinischer Relevanz. Durch die Fortschritte im Bereich der Sequenziertechnologien ist es nun möglich, einerseits ungerichtete molekulare Diagnostiksstrategien aufzubauen, andererseits ganze mikrobiologische Gemeinschaften zu charakterisieren. Solche metagenomischen Projekte verfolgen das Ziel, die Wechselwirkungen zwischen verschiedenen Erregern in einem gemeinsamen Habitat (z. B. gastrointestinale Flora oder Biofilmbildung) besser zu verstehen. Da auch genetische Spuren nicht kultivierbarer Erreger ausgewertet und so umfassende Informationen zur Zusammensetzung verschiedener Probenmatrices in Abhängigkeit verschiedener Parameter (z. B. Probenart und Klimafaktoren) gewonnen werden, können solche metagenomischen Analysen ebenfalls zur Rückverfolgung verdächtiger Proben beitragen.
Die Vollgenomsequenzierung eröffnet eine Vielzahl von neuen Anwendungsgebieten, die auch für wehrwissenschaftliche Fragestellungen bedeutsam sein können: Wie dynamisch verändern sich Krankheitserreger während eines Ausbruchs? Können Genexpressionsstudien die Aussagekraft genotypischer Resistenz- und Virulenzdaten weiter verbessern? Lassen sich neue Therapieoptionen bei multiresistenten Erregern ableiten?
Schneller, höher, weiter
Während sich die bisherigen Projekte vor allem auf bakteriologische Erreger konzentriert haben, wird sich der Fokus in Zukunft auch auf hochpathogene Viren erweitern. Ziel ist es, Arbeitsabläufe weiter zu beschleunigen und die Methode so zu verbessern, dass bereits direkt aus der klinischen Probe ohne vorherige Anzucht spezifische Signaturen hochpathogener Keime samt Virulenz- und Resistenzfaktoren identifiziert werden können. Algorithmen sind zu etablieren, die aus den Vollgenomdaten in silico Typisierungsdaten generieren, um die Rückwärtskompatibilität zu bestehenden Methoden zu gewährleisten. Schließlich wird immer deutlicher, dass viele Fragestellungen nur in Forschungsnetzwerken zufriedenstellend zu beantworten sind. Mit der Universität Münster und der Northern Arizona University (Flagstaff, Arizona, USA) stehen dem Institut für Mikrobiologie der Bundeswehr kompetente Pioniere auf dem Gebiet NGS als Kooperationspartner zur Seite. Der Auf- und Ausbau nationaler, europäischer und internationaler Datenbanken nach festgelegten Qualitätsstandards bleibt weiter eine große Herausforderung. Der neue Funktionsbereich „Mikrobielle Genomik und Bioinformatik“ wird die bisherigen Fähigkeiten des Instituts im Bereich Erregercharakterisierung unter Nutzung neuer Technologien weiter ausbauen und in Verbindung mit der Weiterentwicklung bestehender Datenbanken die Fähigkeit zur Verifizierung einer absichtlichen Freisetzung biologischer Agenzien weiter stärken.
Milzbrand und Heroin
Doch was wurde nun aus den Milzbrandfällen der Drogenkonsumenten? Ergebnisse der Ausbruchsuntersuchungen wurden in Kooperation mit dem Robert-Koch-Institut und weiteren Institutionen auf Fachtagungen der wissenschaftlichen Öffentlichkeit präsentiert. Es zeigte sich, dass die Typisierung der Isolate von 2009/10 und 2012/13 mittels Bestimmung kanonischer SNP-Marker identische Resultate erbrachte. Zusätzlich zeigte auch die der Vaterschaftsanalyse ähnliche Methode MLVA bei 29 von 31 Markern ein identisches Ergebnis. Die Unterschiede liegen dabei in zwei äußerst veränderlichen Regionen, sodass diese Ergebnisse durchaus mit der Theorie vereinbar sind, dass es sich nur um einen „Batch“ an kontaminiertem Heroin handelt. Im Laufe der Untersuchungen wurde allerdings auch ein Stamm mit gleichem Typisierprofil gefunden, der bereits im Jahr 2000 in Norwegen aus einem drogenabhängigen Patienten isoliert wurde. Die Wahrscheinlichkeit, dass auch diese Probe aus dem gleichen „Batch“ stammt, gilt aufgrund der verhältnismäßig langen Zeitspanne als eher unwahrscheinlich. Vollgenomsequenzen zu den Isolaten von 2000, 2009/10 und 2012/13 liegen bei den unterschiedlichsten Forschungsgruppen bereits vor, sind jedoch bislang nur teilweise publiziert [9]. Dadurch konnten die Daten bisher leider nur unzureichend miteinander verglichen werden, um weitere Verbindungen zwischen den Stämmen aufzudecken. Über die letztendliche geographische Herkunft des Stammes kann deshalb momentan nur spekuliert werden, da der spezifische molekulare Fingerabdruck der Ausbruchsstämme bisher in Umweltproben noch nicht beobachtet wurde. Die ähnlichsten bekannten Muster stammen von Isolaten aus dem Gebiet der östlichen Türkei. Im Grenzgebiet zum Iran werden die Haupthandelsrouten für Heroin aus Afghanistan vermutet. Die derzeit populärste Hypothese und wahrscheinlichste Erklärung geht von einer zufälligen Kontamination des Heroins mit Milzbrandsporen während des Transportes entlang der Schmuggelrouten aus.
Interessenkonflikt
Die Autoren erklären, dass im Sinne der Richtlinien des International Commitee of Medical Journal Editors keine Interessenskonflikte bestehen.
Literatur
- Radun D et al. (2010): Preliminary case report of fatal anthrax in an injecting drug user in North-Rhine-Westphalia, Germany, December 2009. Euro Surveill. 2010 Jan 14; 15(2).
- ECDC (2009): Joint ECDC and EMCDDA Threat Assessment. Anthrax outbreak in drug users, Scotland. 21 December 2009.
- Holzmann T et al. (2012): Fatal anthrax infection in a heroin user from southern Germany, June 2012. Euro Surveill. 2012 Jun 28; 17(26).
- Rothberg JM et al. (2011): An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011 Jul 20; 475(7356): 348-52.
- Mellmann A et al. (2011): Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One. 2011; 6(7):e22751.
- Mellmann A et al. (2006): DNA-Sequenz-basiertes Frühwarnsystem für den Nachweis von MRSA-Ausbrüchen. Laborwelt, Nr. 1/2006.
- Hoffmann B et al. (2011): Novel Orthobunyavirus in Cattle, Europe, 2011. Emerg Infect Dis. 2012 March; 18(3): 469 – 472.
- Robert-Koch-Institut (2011): Abschlussbericht zum EHEC/HUS-Ausbruch: Abschließende Darstellung und Bewertung der epidemiologischen Erkenntnisse im EHEC O104:H4 Ausbruch Deutschland 2011.
- Rückert C et al. (2012): Draft genome sequence of Bacillus anthracis UR-1, isolated from a German heroin user. J Bacteriol. 2012 Nov; 194(21):5997 - 8.
Bildquellen:
Abbildungen 1 und 2: Grafiken erstellt durch Institut für Mikrobiologie der Bundeswehr, unter freundlich genehmigter Verwendung von Geräteabbildungen der Firmen Dell Computer, Life Technologies und Thermo Scientific, alle Deutschland.
Abbildung 2: Gruppenfoto: Institut für Mikrobiologie der Bundeswehr.
Datum: 06.03.2014
Quelle: Wehrmedizinische Monatsschrift 2014/2










{kind=link}
{kind=link}