EINSATZ MOLEKULARER TYPISIERUNGSMETHODEN ZUR UNTERSUCHUNG VON Q-FIEBERAUSBRÜCHEN
Application of molecular Typing Methods in Investigations of Q fever Outbreaks
Aus dem Institut für Mikrobiologie der Bundeswehr, München (Leiter: Oberstarzt Prof. Dr. L. Zöller)
Dimitrios Frangoulidis
Die Zoonose Q-Fieber (Erreger: Coxiella burnetii) kommt weltweit und damit auch in Einsatzgebieten der Bundeswehr vor. Diese ubiquitäre Verfügbarkeit, die Übertragbarkeit als Aerosol und die extrem niedrige Infektionsdosis waren Gründe für die Verwendung und Erforschung von Coxiella (C.) burnetii in den B-Waffen-Programmen während des Kalten Krieges. Auch gegenwärtig gilt der Erreger weiterhin als potenzielles bioterroristisches Agens.
Regelmäßig werden in Endemiegebieten des Q-Fiebers natürliche Erkrankungsausbrüche beschrieben, die fast immer mit Abortgeschehen in der entsprechenden Tierpopulation wie Schafe oder Ziegen in Verbindung gebracht werden können. Damit unterliegen Bundeswehrkontingente beim Einsatz in solchen Naturherden einem besonderen natürlichen wie auch artifiziellen Infektionsrisiko. Daher sind sogenannte „bioforensische“ Untersuchungen zur molekularen Typisierung des Erregers erforderlich, um im Falle eines Krankheitsausbruchs eine kausale Rückverfolgung und Unterscheidung zwischen natürlicher und gezielter Infektion zu ermöglichen.
Methoden:
Literaturrecherche und Auswertung eigener Etablierungsarbeiten zur Entwicklung und Bewertung von Anwendbarkeit und Qualität molekularbiologischer Typisierungsmethoden für C. burnetii.
Ergebnisse:
Aktuell verwendete Methoden erreichen eine Zuordnung von Stämmen/Proben zu bestimmten Regionen, können jedoch noch nicht sicher Einzelstämme unterscheiden. Die Verfahren weisen außerdem einen unterschiedlichen Grad an Standardisierung und Harmonisierung auf.
Schlussfolgerungen:
Zurzeit ist eine Kombination verschiedener Typisierungsmethoden für Coxiella burnetii am besten geeignet, um sowohl ein hohes Diskriminierungsniveau zu erreichen, als auch epidemiologische Zusammenhänge zu erfassen und damit eine bioforensische Analyse von Ausbruchsmaterialien zu ermöglichen. Zukünftig sind eine ausreichende Standardisierung der Verfahren, die Etablierung internationaler Typisierungsdatenbanken und Verfeinerungen in der Einzelstammanalyse anzustreben.
Summary
Background:
The worldwide distributed zoonosis Q fever (pathogen: Coxiella burnetii) is transmissible via aerosol and shows an extreme low infectious dose. These features were the reason beyond others including this pathogen in the biological warfare program of the cold war era. Still Coxiella (C.) burnetii is an agent with a bioterrorist potential. Endemic Q fever outbreaks, occurring periodically, often with hundreds of affected people, mainly base on abortion situations in ruminant flocks like sheep and goats. This endemic disease is also seen in regions of the Bundeswehr missions, causing outbreaks which may affect soldiers. Here we present different bio-forensic tools used for trace back analysis in outbreaks as well as to differentiate between natural and artificial based situations.
Methods:
Using published literature and own data from method evaluations all known genomic based analysis methods for typing of C. burnetii are presented and evaluated due to their reliability and quality.
Results:
Today used methods could allocate strains/probes to distinct geographical regions, but are failing to differentiate on a single isolate level. In addition the assays are showing different levels due to standardisation and harmonisation.
Conclusions:
To gain a high discriminatory level and to analyse epidemiological relations for bioforensic work, combinations of different molecular typing systems are necessary when looking on outbreak derived material. Standardisation of methods, establishment of international used databases and a further progress in resolution of the techniques are the challenges for the future.
1. Einführung
Die Zoonose Q-Fieber, ausgelöst durch das gramnegative Bakterium Coxiella burnetii, ist seit ihrer Erstbeschreibung 1937 in fast jedem Land der Welt dokumentiert worden. Der Erreger löst bei den betroffenen Personen in gut 50 % der Fälle eine akute, grippeähnliche Symptomatik aus, die oft mit einer atypischen Pneumonie und/oder mit einer akuten Hepatitis vergesellschaftet ist. Besonders gefürchtet sind chronische Erkrankungsverläufe (weniger als 2 % aller Infektionen). Diese erfordern im Gegensatz zu der gut behandelbaren akuten Form langjährige Antibiotikagaben und gehen zumeist mit einer chronischen Entzündung der Herzklappen einher. Infolge der daraus resultierenden Komplikationen (Klappenersatz, Herzrhythmusstörungen) liegt die langfristige Überlebensrate des chronischen Q-Fiebers nur bei etwa 70 - 75 % [1]. Aufgrund der weltweiten Verfügbarkeit, hohen Umweltstabilität, Übertragbarkeit als Aerosol und extrem niedrigen aerogenen Infektionsdosis (< 10 Erreger) war Coxiella (C.) burnetii während des Kalten Krieges frühzeitig Bestandteil der staatlichen Forschungsprogramme in den Vereinigten Staaten und der ehemaligen Sowjetunion zur Entwicklung von biologischen Kampfstoffen. Aber auch nach Beendigung dieser Programme wird dem Erreger wegen der genannten Eigenschaften ein bioterroristisches Potenzial durch die Centers for Disease Control and Prevention (CDC) in Atlanta beigemessen [2]. Weltweit werden regelmäßig natürlich auftretende Q-Fieberausbrüche mit zum Teil mehreren Hundert betroffenen Personen beschrieben, die fast immer mit Abortgeschehen in der entsprechenden Tierpopulation, zum Beispiel Schafe und Ziegen, in Verbindung stehen [3-5]. Auch in den meisten bisherigen Einsatzgebieten der Bundeswehr ist diese Erkrankung endemisch und löst regelmäßig entsprechende Erkrankungen aus, die zum Teil auch das militärische Personal betreffen können [6; 7]. Bei einem Ausbruchsgeschehen mit einem Erreger, der ein bioterroristisches Potenzial besitzt, muss immer geprüft werden, ob es Hinweise auf eine nicht natürlich bedingte Ursache gibt. Neben der Bewertung epidemiologischer Zusammenhänge wird in den letzten Jahren auch verstärkt das Erregergenom untersucht, um diese fachlich und politisch bedeutsame Fragestellung zu klären. Die Aufklärung der Milzbrandbriefattacken von 2001 in den USA haben gezeigt, wie wichtig, aber auch komplex solche gerichtsverwertbaren Untersuchungen sein können. Im Rahmen dieser langjährigen Studien rückte ein relativ junger Zweig der Biowissenschaften in die öffentliche Wahrnehmung, die sogenannte mikrobielle Forensik (kurz: Bioforensik). Deren Ziel es ist es, möglichst zweifelsfrei Infektketten aufzuklären und Erkrankungen einer exakten Quelle beziehungsweise Ursache zuordnen zu können. Die Bedeutung dieses Wissenschaftszweiges wurde erst im Sommer letzten Jahres eindrucksvoll unter Beweis gestellt, als während des bisher größten EHEC (Enterohämorrhagische Escherichia Coli) -Ausbruchs in Deutschland mit schwersten Nierenschädigungen (Hämolytisch- Urämisches Syndrom) mit modernsten molekularbiologischen Methoden nach dem ursächlichen Erreger im Rahmen der epidemiologischen Herdanalyse gesucht wurde [8].
In dem hier vorliegenden Artikel soll nun kurz die Entwicklung der Methoden zur molekularbiologischen Typisierung und Differenzierung von C. burnetii geschildert und deren Qualität und Anwendbarkeit für die Bioforensik bewertet werden.
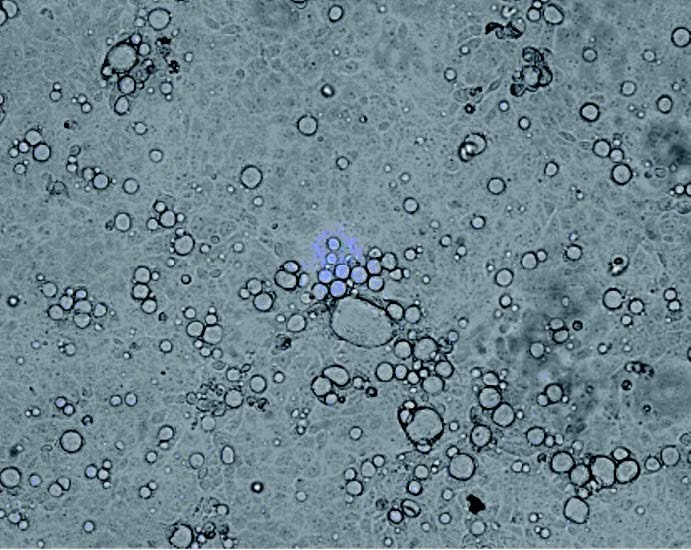
Abb 1: Mit Coxiella burnetii infizierte BGMZellkultur bildet typische Vakuolen als Zeichen der Infektion. (Foto: Institut für Mikrobologie der Bundeswehr)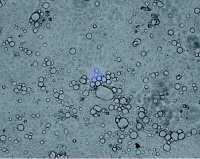
2. Ergebnisse
Für die epidemiologischen Analysen wurden verschiedenste Methoden zur Genotypisierung von C. burnetii entwickelt.
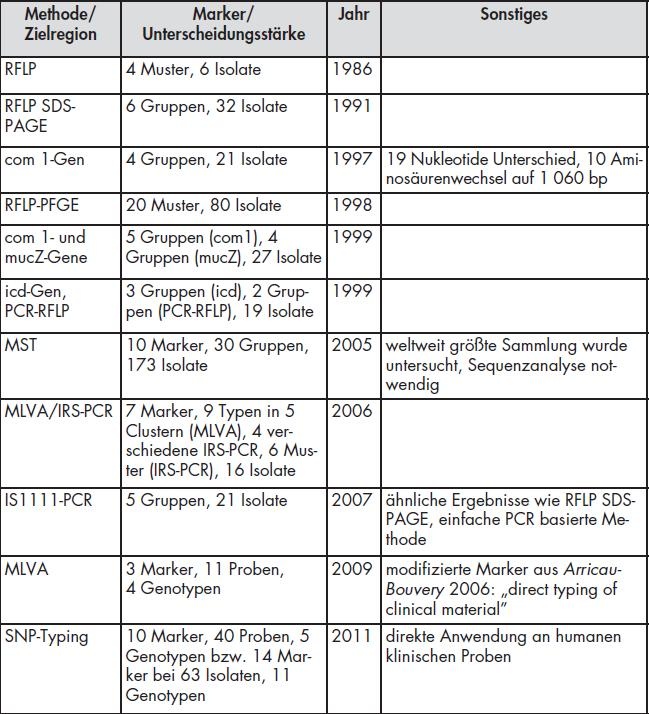
Initiale Verfahren (vor 2005) basierten auf dem Nachweis von Coxiellen-spezifischen Plasmiden, auf Restriktionsfragmentanalysen (RFLP) mit Pulsfeldgelelektrophorese (RFLP) und Untersuchungen von sogenannten „house-keeping“ Genen wie mucZ, com1 und icd [9-12]. Alle Methoden konnten zwar gewisse Aussagen über die epidemiologische Verteilung von Stämmen und eine grobe Differenzierung von Isolaten ermöglichen, zeigten jedoch große Probleme bei der Reproduzierbarkeit von Ergebnissen innerhalb des Labors und auch bei der Vergleichbarkeit von Resultaten aus verschiedenen Institutionen.
2.1 Verfahren vor 2005
2.1.1 Plasmidtypbestimmung
C. burnetii kann fünf verschiedene Plasmidvarianten besitzen: (QpH1, QpRS, QpDV, QpDG) und enthält plasmidhomologe Sequenzen im Chromosom bei einer Isolatgruppe [13-15]. Mithilfe dieser Plasmide war eine Klassifizierung der Coxiellen in fünf genomische Gruppen möglich, die zum Teil auch bestimmte regionale Zusammenhänge erfassen konnte. Eine zuerst erhobene Vermutung, dass bestimmte Plasmide bevorzugt bei akuten beziehungsweise chronischen Erkrankungsverläufen vorkommen, konnte letztendlich nicht bestätigt werden [16].
2.1.2 RFLP-Analyse und SDS-PFGE
Die Restriktions-Fragment-Längen-Polymorphismus (RFPL)-Analyse mittels SDSPulsfeldgelektrophorese (SDS-PFGE) mit anschließendem Enzymverdau der DNA führte Anfang der 90er Jahre zu einer Einteilung von 6 Gruppen, die eine hohe Übereinstimmung mit der Plasmidtypisierung aufwies [9]. Eine weitere Verfeinerung der Methode erbrachte sogar den Nachweis von 20 verschiedenen Mustern bei der Untersuchung einer Stammsammlung mit Isolaten aus Europa, den USA, Asien und Afrika [10].
2.1.3 Nachweis von com1-, mucZ- und icd-Genen
Ab 1997 untersuchten verschiedene Arbeitsgruppen die Sequenzvariationen dieser Coxiellengene in der Hoffnung, mehr Aufschluss über die klinische Bedeutung dieser Varianten zu erhalten. Letztendlich konnten jedoch mit diesen Verfahren weder genaue regionale Zuordnungen noch Hinweise auf bestimmte Krankheitsverläufe (akut/chronisch) erzielt und somit auch keine verwertbaren epidemiologischen Untersuchungen durchgeführt werden [11-12].
2.2 Hoch auflösende Verfahren nach 2005
Mit der Verfügbarkeit von Gesamtgenomdaten (ab 2003) wurde eine ganze Reihe sehr diskriminierender Verfahren wie die „Multispacer Sequence Typing“ (MST)-Methode, die Verteilung von IS1111-Elementen, die Bestimmung von „Multiple loci variable number of tandem repeats“ (MLVA/VNTR) und die Identifizierung von „Single Nucleotid Polymorphisms (SNP)“ entwickelt [17-19].
2.2.1 “Multispacer Sequence Typing” (MST)
Damit werden die Regionen zwischen einzelnen Genen auf Sequenzvariationen hin untersucht, da hier der sogenannte Selektionsdruck auf funktionelle Gene geringer ist. Bei der Entwicklung des Verfahrens 2005 konnten 173 C. burnetii-Stämme aus der ganzen Welt in 30 verschiedene Untergruppen eingeteilt werden [17], die auch bestimmten geografischen Regionen und den oben erwähnten verschiedenen Plasmidtypen zuzuordnen waren. Diese deutlich verbesserte Auflösungsstärke, verbunden mit der eindeutigen, konstanten und vergleichbaren Bestimmungssicherheit der genomischen Marker, stellte eine deutliche Verbesserung gegenüber den früheren Gel-basierten Verfahren dar. Somit waren erstmals Vergleichsuntersuchungen zwischen verschiedenen Laboren möglich und auch die eindeutige Zuordnung von neuen Isolaten zu bestehenden Gruppierungen. Mittlerweile existiert eine internetbasierte Datenbank für MST, die weltweit zur Typisierung von C. burnetii genutzt werden kann (www.ifr48.timone.univ-mrs.fr/MST_Coxiella/mst).
2.2.2 “Variable number of tandem repeats” (VNTR)
Diese direkten Wiederholungen von Genombasen (Nukleotide), die auch Minisatelliten genannt werden, bestehen aus bis zu 100 Nukleotiden. Die Varianz in der Anzahl der Wiederholungen kann zur Unterscheidung von DNA-Proben herangezogen werden. Diese Varianz kann so spezifisch sein, dass eine sichere Unterscheidung beziehungsweise Zuordnung von zwei Proben möglich ist. Ursprünglich bei der humanen Genetik für die forensische Untersuchung von DNA-Proben entwickelt („genetischer Fingerabdruck“ zum Beispiel im Rahmen des Vaterschaftsnachweises), dient dieses Verfahren zunehmend häufiger dazu, Bakterienstämme zu differenzieren.
2006 wurde ein entsprechendes Verfahren für die Untersuchung von Coxiellen sowohl in Frankreich in Zusammenarbeit mit unserem Institut als auch unabhängig davon in Holland entwickelt [18, 20]. Bis zu 17 verschiedene Genregionen werden auf ihre spezifische Varianz hin untersucht und erlauben eine sehr gute Diskriminierung von Isolaten. Einer Sammlung von 42 Stämmen konnten 37 verschiedene Genotypen zugeordnet werden. Allerdings bestehen Probleme bei der Vergleichbarkeit von Ergebnissen zwischen verschiedenen Laboren. Dies ist neben der noch fehlenden Abstimmung über eine einheitliche Nomenklatur besonders den unterschiedlichen Geräteplattformen zur Längenbestimmung der “tandem repeats”- PCR-Produkte geschuldet. Eine Variante dieser Methode mit drei ausgewählten modifizierten Markern wurde darüber hinaus erfolgreich 2009 bei der Aufklärung eines massiven QFieberausbruchs in den Niederlanden angewandt. Hierbei gelang sogar erstmals eine direkte Typisierung aus humanen klinischen Untersuchungsmaterialien, wobei sich vier verschiedene Genotypen identifizieren ließen [21]. Wie bei der MST-Technik existiert eine internetbasierte Datenbank, die für die Auswertung von Typisierungsergebnissen verwendet werden kann (http://minisatellites.u-psud.fr/).
2.2.3 IS1111-Typisierung
Die Coxiella-spezifische IS1111-Insertionssequenz, die für eine Transposase kodiert, kommt bis zu 56-mal im Genom vor. Sie ist daher der klassische Genort, der in molekularbiologischen Nachweisverfahren wie der Polymerase Kettenreaktion (PCR) verwendet wird. 2007 fand eine amerikanische Arbeitsgruppe heraus, dass die der Genregion vorgelagerten Sequenzanteile sich untereinander unterscheiden können und sich so für Typisierungsansätze eignen [22]. Mit Hilfe von 5 Markern konnten somit fünf verschiedene Genotypengruppen ermittelt werden, die ebenfalls eine hohe Übereinstimmung mit den bereits bekannten Ergebnissen der plasmidbasierten Systeme zeigten. Da dieses Verfahren jedoch nur auf der einfachen Auswertung von PCR-Reaktionen basiert, ist es ohne größeren technischen und organisatorischen Aufwand in jedem Labor etablierbar und leicht anzuwenden. Eine hohe Vergleichbarkeit der erzielten Ergebnisse ist damit gewährleistet. Unser Institut hat das ursprünglich fünf Marker umfassende System auf 32 Marker erweitert und konnte so eine deutlich höhere Auflösungsstärke erzielen. Erstmals ist es mit diesem modifizierten Verfahren möglich, verschiedene Stämme zu unterscheiden und so nahezu eine Identifizierung von Einzelisolaten zu erzielen.
2.2.4 Single Nucleotide Polymorphism (SNP)-Typing
Dieses Verfahren, das in den letzten Jahren zunehmend breiter bei der genomischen Analyse von Bakterien genutzt wird, wurde erstmals von einer holländischen Arbeitsgruppe 2011 bei C. burnetii angewandt [23]. Im selben Jahr wurde eine erweiterte Variante mit mehr SNP-Positionen beschrieben, die eine Diskriminierungsstärke vergleichbar mit der MST-Methode zeigte (s. 2.2.1). 40 Isolate einer Sammlung konnten in acht verschiedene Genotypen unterteilt werden. Dabei korrelierten diese mit dem geografischen Ursprung der Proben, dem klinischen Erscheinungsbild und dem Plasmidtyp der Proben [19]. Ähnlich zur IS1111- Methode, sind auch hier entsprechende Sequenzveränderungen leicht, sicher und eindeutig zu identifizieren. Damit ist diese Methode sehr gut standardisierbar und in verschiedenen Laboren umsetzbar. Auch besteht die Möglichkeit, viele Marker gleichzeitig auf entsprechende Hochdurchsatzplattformen umzusetzen und somit eine schnelle und genaue Zuordnung von Isolaten durchzuführen.
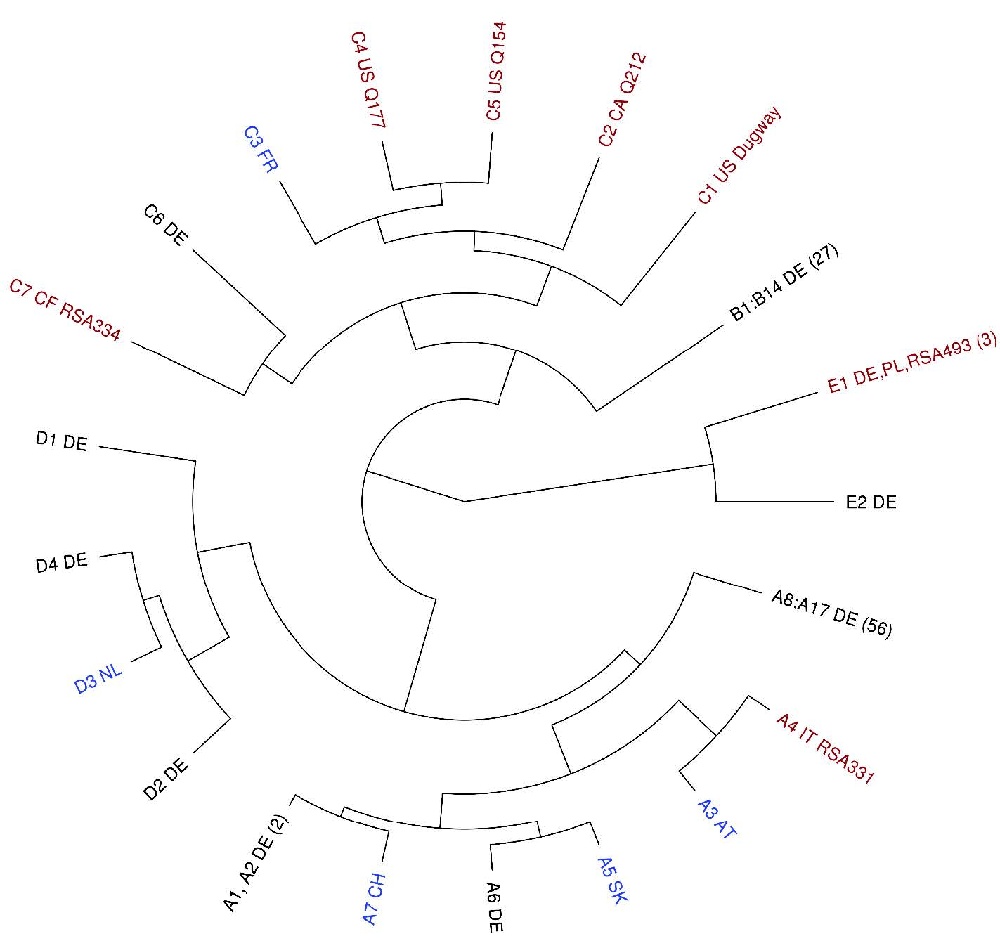
Abb 2: Darstellung der MLVA-Typisierungsergebnisse mittels der sogenannten Neighbor-Joining- Tree-Methode. Die einzelnen Länder-„Cluster“ sind dabei gut zu unterscheiden.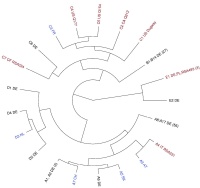
3. Diskussion
Eine Übersicht der wichtigsten hier vorgestellten Verfahren und ihrer Eigenschaften wird noch einmal in Tabelle 1 gegeben. Die Verfahren vor 2005 zeigen hierbei neben der zum Teil sehr geringen Auflösungsstärke auch deutliche Schwächen bei der Reproduzierbarkeit der Ergebnisse und der Etablierbarkeit der Methode in anderen Laboratorien. Die neueren Systeme nach 2005 weisen überwiegend sehr gute diskriminierende Ergebnisse (MLVA, IS1111) auf, können aber auch bei der Analyse epidemiologischer Zusammenhänge hilfreich sein (SNP, MST). Mit PCR-basierten Systemen wie SNP und IS1111 ergaben sich dabei verständlicherweise weniger Probleme in der Etablierung der Verfahren im Laboralltag. Sie sind darüber hinaus auch recht kostengünstig. Verfahren, die eine partielle Genomsequenzanalyse voraussetzen wie MST und MLVA erfordern einen deutlich höheren Arbeitsund Analyseaufwand und mitunter auch eine relevante Mehrarbeit zum Abgleich der zugrunde liegenden Nomenklatur beziehungsweise Auswertealgorithmen. Regionale Zuordnungen auf Länder- oder Kontinentebene und zum Teil auch auf kleinere geografische Strukturen wie Bundesländer und Landkreise (mittels MLVA) sind möglich. Die Einzelstammunterscheidung ist noch nicht komplett realisierbar, aber das erweiterte IS1111-Verfahren zeigte von allen untersuchten Methoden die höchste Auflösungsstärke.
Die Anforderungen an molekularbiologische Typisierungsverfahren in der Mikrobiologie sind vielfältig. Oberstes Ziel ist es, einem Erkrankungsfall/Ausbruchsgeschehen eine eindeutige Ursache zuordnen zu können. Durch die Primärdiagnostik wird der ursächliche Erreger identifiziert, um neben der Auswahl der richtigen Therapie auch gegebenenfalls notwendige epidemiologische Aspekte einer Infektion rechtzeitig berücksichtigen zu können. Dies ist zum Beispiel bei einer von Mensch zu Mensch übertragbaren Erkrankung besonders wichtig, damit eine weitere Ausbreitung möglichst frühzeitig gestoppt werden kann. In zweiter Linie und oft schon parallel zur Primärdia - gnostik findet aber auch sehr häufig eine weitere Differenzierung nach phäno- und genotypischen Merkmalen des isolierten Erregers statt. Bei der Phänotypie ist die Ermittlung des antimikrobiellen Resistenzverhaltens am häufig - sten. Dieses seit gut einem halben Jahrhundert angewandte Verfahren wird in den letzten 15 Jahren aber zunehmend durch genotypische Untersuchungsverfahren ergänzt oder teilweise ersetzt. So gibt es zum Beispiel bei Mycobacterium tuberculosis und Staphylococcus aureus mittlerweile molekularbiologische Schnellnachweisverfahren, die binnen zwei bis drei Stunden therapeutisch relevante Antibiotikaresistenzen identifizieren können. Auch die Rückverfolgung von Krankheitserregern zu einer Infektionsquelle gewinnt zunehmend an Bedeutung, allein schon aus dem einfachen Grund, um diese Ursache zu beseitigen und damit weitere Infektionen zu vermeiden.
Die dabei verwendeten Methoden müssen auf der einen Seite eine zweifelsfreie Zuordnung eines Ausbruchsstamms zu einer vermuteten Quelle ermöglichen, darüber hinaus auch möglichst schnell ein Ergebnis zeigen und dabei in der Durchführung und Auswertung einfach, praktikabel und sicher sein. Diese an sich schon hohen Anforderungen an die Bioforensik werden im militärischen Bereich noch einmal verschärft, da hierbei durch die gewonnenen Ergebnisse weitreichende taktische Entscheidungen getroffen werden können.
Coxiella burnetii bedingt durch seine weite Verbreitung, seine luftgebundene Übertragbarkeit über mehrere Kilometer hinweg und seine extrem niedrige Infektionsdosis selten Einzelerkrankungen sondern fast immer Gruppengeschehen. Durch die Inkubationszeit von 2 bis 4 Wochen, den eher unspezifischen Symptombeginn und die große Bandbreite an klinischen Verläufen ist die Diagnosestellung sehr häufig verzögert. Dies stellt noch höhere Anforderungen an eine Rückverfolgung eines Erkrankungsausbruchs, da zwischen Diagnosesicherung und Infektionsbeginn manchmal mehrere Wochen liegen können. Coxiellen weisen jedoch eine extrem hohe Umweltstabilität auf und können damit noch nach Monaten in der Umwelt nachgewiesen und gelegentlich mittels spezialisierter Zellkulturverfahren angezüchtet werden. Da bei einem natürlich bedingten Infektionsgeschehen immer eine entsprechend befallene Tierherde (zumeist Schafe oder Ziegen) als Ursache in Frage kommt und infizierte Tiere den Erreger dauerhaft ausscheiden können, ist die veterinärmedizinische Beprobung solcher Bestände unerlässlich. Im Gegensatz zu menschlichen Untersuchungsproben finden sich hier zumeist auch sehr große Erregermengen, die entsprechende diagnostische und forensische Untersuchungen gut ermöglichen. Beim Menschen ist die DNA des Erregers nur etwa zwei Wochen nach Symptombeginn nachweisbar. Danach erfolgt die Diagnosesicherung durch die Bestimmung erregerspezifischer Antikörper. Außerdem ist die zirkulierende DNA-Menge im peripheren Blut sehr gering. Untersuchungen aus den Niederlanden der letzten Jahre zeigten aber auch, dass es prinzipiell möglich ist, aus Proben, die bei akut an Q-Fieber erkrankten Patienten gewonnen wurden, nicht nur die DNA des Erregers nachzuweisen, sondern sogar auch eine molekularbiologische Typisierung durchzuführen [23]. So gelang es damit auch, den bisher größten und langandauerndsten Q-Fieberausbruch weltweit (2007 bis 2011 mit über 4 000 Erkrankten) einer Infektionsquelle exakt zuzuordnen. Die in Holland betriebene Massentierhaltung von Ziegen – vergleichbar der Milchkuhwirtschaft in Deutschland – in direkter Nähe zu dicht bewohnten Regionen wurde eindeutig als Ursache des Ausbruchs identifiziert. Wahrscheinlich über einen längeren Zeitraum hinweg waren die meisten Ziegenbestände mit C. burnetii infiziert worden und gaben während der saisonalen Lammungsperiode extrem große Mengen des Erregers an die Umwelt ab. Die landestypischen Windverhältnisse trugen dann weiter dazu bei, dass diese Erregerlast über eine große bewohnte Fläche verteilt wurde und somit die enorme Anzahl an Erkrankten resultierte.
In diesem Artikel werden die Stärken und Schwächen der vorgestellten Verfahren zur molekularbiologischen Typisierung von C. burnetii aufgezeigt, die für die Auswertung und Verwendung der erzielten Ergebnisse bedeutsam sind. Eine zu geringe Auflösungsstärke wie bei der MST oder beim SNP-Typing erlaubt im Ausbruchsgeschehen keine sichere Zuordnung zu einer vermuteten Quelle, kann aber größere regionale Zusammenhänge erfassen. Dagegen kann eine Diskriminierung auf Stammebene mittels IS1111-Analyse oder MLVA eine sichere Verwandtschaft zwischen Isolaten belegen, aber epidemiologisch nur eingeschränkt verwertet werden. Zusätzlich können im Ausbruchsgeschehen mitunter verschiedene Varianten eines Erregers auftreten oder sich während eines Wirtswechsels oder einer Infektion entwickeln und damit ein komplexes Muster an verschiedenen Genotypen bilden. Unabhängig davon ist aber für alle Verfahren eine eindeutige und übertragbare Nomenklatur und Stabilität der ermittelten Ergebnisse unabdingbar, da sonst falsche beziehungsweise variable Ergebnisse und Interpretationen entstehen können. Im Rahmen der Spezialdiagnostik und medizinischen B-Aufklärung am InstMikroBioBw ist neben der möglichst schnellen und sicheren vorläufigen Erregeridentifizierung in einem Ausbruchsgebiet auch zunehmend der Anspruch vorhanden, eine vorläufige Zuordnung des Erregers zu ermöglichen. Dies stellt natürlich eine zusätzliche enorme Herausforderung an die eventuell eingesetzten Verfahren und technischen Plattformen einschließlich des Personals vor Ort dar.
Alle Erfahrungen der letzten Jahre haben zudem sehr deutlich gemacht, dass die Typisierung von Krankheitserregern auch eine internationale Herausforderung ist, um möglichst viele auftretende Erregervarianten erfassen und auswerten zu können. Die zunehmende Verbreitung dieser internetbasierten Datenbanken verlangt aber von allen beteiligten Institutionen eine hohe Disziplin beim Erstellen, Bearbeiten und Entwickeln solcher Plattformen. Auch muss hierbei eine gewisse Bereitschaft vorhanden sein, Daten und Wissen zu teilen. Dies führt in gewisser Weise natürlich zu einem Konflikt beim Datenschutz und bei der Datensicherheit. Trotzdem wird an einem geeigneten Maß an Öffentlichkeit der ermittelten Typisierungsdaten kein Weg vorbeiführen.
Tab 1: Zusammenfassung der wichtigsten Typisierungsmethoden für Coxiella burnetii.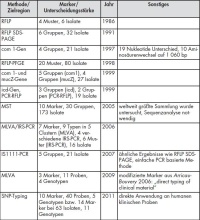
5. Schlussfolgerungen
Zur Zeit ist eine Kombination von verschiedenen molekularbiologischen Typisierungsmethoden für Coxiella burnetii am besten geeignet, sowohl ein hohes Diskriminierungsniveau zu erreichen als auch epidemiologische Zusammenhänge zu erfassen und damit eine bioforensische Analyse von Ausbruchsmaterialien zu ermöglichen. Die Verfahren MST, IS-1111 und MLVA sind am InstMikroBioBw in München etabliert und werden seit Jahren verwendet und weiterentwickelt. Das seit 2011 verfügbare SNP-Typing wird aktuell auf seine Eignung und Umsetzbarkeit geprüft. Dabei wurden bereits aussagekräftige Ergebnisse mit einer modifizierten Nachweistechnik erzielt. Ähnlich wie von der holländischen Arbeitsgruppe gezeigt, kann damit auch der Sanitätsdienst der Bundeswehr einen Q-Fieberausbruch zu einer Quelle zurückverfolgen und eine recht gesicherte Aussage über die Herkunft oder Abstammung einer Probe treffen. Eine Verlagerung dieser Techniken in die Vor-Ort-Analyse ist aber zurzeit noch nicht möglich beziehungsweise noch nicht zufriedenstellend evaluiert. Neben diesem Aspekt sind eine ausreichende Standardisierung der Verfahren, die Etablierung internationaler Typisierungsdatenbanken und weitere Verfeinerungen in der Einzelstammanalyse die zukünftigen Herausforderungen auf diesem Arbeitsgebiet.
Danksagung
Der Autor möchte sich ganz herzlich bei allen ehemaligen und aktuellen Mitarbeitern der TE 040 „Medizinische Spezialdiagnostik/Hochsicherheitslabor“, ihrem Leiter, Oberstveterinär Prof. Dr. H. Meyer, am Institut für Mikrobiologie der Bundeswehr für ihre langjährige und unermüdliche Unterstützung bedanken. Besonderer Dank gilt Frau Claudia Kahlhofer, Frau Hauptfeldwebel Anke Stark, Herrn Regierungsrat Markus Antwerpen und Herrn Stabsveterinär Dr. Matthias Hanczaruk, die mit viel Engagement, Enthusiasmus und stetiger Hilfsbereitschaft dazu beigetragen haben, den Schwerpunkt Coxiella burnetii/Q-Fieber am Institut zu etablieren und auszubauen.
Literatur
- Tissot-Dupont H, Raoult D: Q fever. Infect Dis Clin North Am 2008; 22(3): 505-14, ix. Review.
- Bioterrorism Agents/Diseases. In: Emergency Preparedness and Response, Centers for Disease Control and Prevention: http://www.bt.cdc.gov/agent/agentlistcategory. asp.
- Porten K, Rissland J, Tigges A, et al. : A super- spreading ewe infects hundreds with Q fever at a farmers’ market in Germany.BMC Infect Dis 2006; 6(6): 147.
- Gilsdorf A, Kroh C, Grimm S, Jensen E, Wagner-Wiening C, Alpers K: Large Q fever outbreak due to sheep farming near residential areas, Germany, 2005. Epidemiol Infect 2008;136(8): 1084-1087. Epub 2007 Sep 25.
- Dijkstra F, Hoek W, Wijers N, et al.: The 2007-2010 Q fever epidemic in the Netherlands: characteristics of notified acute Q fever patients and the association with dairy goat farming. FEMS Immunol Med Microbiol 2011; Oct 25. doi: 10.1111/j.1574-695X.2011.00876.x .
- Faas A, Engeler A, Zimmermann A, Zöller L: Outbreak of Query fever among Argentinean special police unit officers during a United Nations mission in Prizren, South Kosovo.Mil Med 2007; 172(10): 1103- 1106.
- Faix DJ, Harrison DJ, Riddle MS, et al.: Outbreak of Q fever among US military in western Iraq, June-July 2005.Clin Infect Dis 2008 ;46(7): e65-68.
- Mellmann A, Harmsen D, Cummings CA, et al.: Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One. 2011; 6(7): e22751. Epub 2011 Jul 20.
- Hendrix L, Samuel J, Mallavia L: Differentiation of Coxiella burnetii isolates by analysis of restriction-endonuclease-digested DNA separated by SDS-PAGE. J Gen Microbiol 1991; 137: 269-276.
- Jäger C, Willems H, Thiele D, Baljer G: Molecular characterization of Coxiella burnetii isolates. Epidemiol Infect 1998; 120(2): 157-164.
- Nguyen SV, Hirai K: Differentiation of Coxiella burnetii isolates by sequence determination and PCR-restriction fragment length polymorphism analysis of isocitrate dehydrogenase gene. FEMS Microbiol Lett 1999; 180(2): 249-254.
- Sekeyová Z, Roux V, Raoult D: Intraspecies diversity of Coxiella burnetii as revealed by com1 and mucZ sequence comparison. FEMS Microbiol Lett 1999; 180(1): 61-67.
- Jäger C, Lautenschläger S, Willems H, Baljer G: Coxiella burnetii plasmid types QpDG and QpH1 are closely related and likely identical. Vet Microbiol 2002; 89(2-3): 161-166.
- Lautenschläger S, Willems H, Jäger C, Baljer G: Sequencing and characterization of the cryptic plasmid QpRS from Coxiella burnetii. Plasmid 2000; 44(1): 85-88.
- Willems H, Ritter M, Jäger C, Thiele D: Plasmid-homologous sequences in the chromosome of plasmidless Coxiella burnetii Scurry Q217. J Bacteriol 1997; 179; (10): 3293-3297.
- Thiele D, Willems H: Is plasmid based differentiation of Coxiella burnetii in 'acute' and 'chronic' isolates still valid? Eur J Epidemiol 1994; 10(4): 427-434.
- Glazunova O, Roux V, Freylikman O,et al. : Coxiella burnetii genotyping. Emerg Infect Dis 2005; 11(8): 1211-1217.
- Arricau-Bouvery N, Hauck Y, Bejaoui A, et al.: Molecular characterization of Coxiella burnetii isolates by infrequent restriction site-PCR and MLVA typing. BMC Microbiol 2006; 6(26): 38.
- Hornstra HM, Priestley RA, Georgia SM, et al.: Rapid typing of Coxiella burnetii. PLoS One. 2011;6(11):e26201. Epub 2011 Nov 2.
- Svraka S, Toman R, Skultety L, Slaba K, Homan WL: Establishment of a genotyping scheme for Coxiella burnetii. FEMS Microbiol Lett 2006; 254: 268-274.
- Klaassen CH, Nabuurs-Franssen MH, Tilburg JJ, Hamans MA, Horrevorts AM: Multigenotype Q fever outbreak, the Netherlands. Emerg Infect Dis 2009; 15(4): 613-4.
- Denison A, Thompson H, Massung R: IS1111 insertion sequences of Coxiella burnetii: characterization and use for repetitive element PCR-based differentiation of Coxiella burnetii isolates. BMC Microbiol 2007; 7: 91.
- Huijsmans CJ, Schellekens JJ, Wever PC, et al.: Single-nucleotide-polymorphism genotyping of Coxiella burnetii during a Q fever outbreak in The Netherlands. Appl Environ Microbiol. 2011 Mar;77(6):2051-7. Epub 2011 Jan 21.
Datum: 28.08.2012
Quelle: Wehrmedizinische Monatsschrift 2012/4








{kind=link}
{kind=link}
{kind=link}