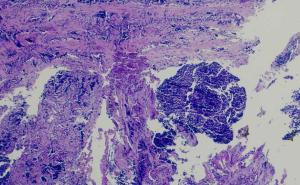
Die Kenntnis auch von selten vorkommenden Tumorläsionen und ihres biologischen Verhaltens ist von Bedeutung beim weiteren diagnostischen und therapeutischen Vorgehen. Häufig sind in derartig selten auftretenden Krankheitsfällen eine spezielle diagnostische Methodik und ein interdisziplinäres Zusammenwirken erforderlich.
Fallbericht 1
Cholesteatom des Nierenbeckens
Eine 61-jährige Patientin kam unter der klinischen Diagnose einer chronischen obstruktiven Bronchitis und Pansinusitis bei akuter Verschlechterung der Beschwerden mit Husten, Luftnot und purulentem Auswurf zur stationären Aufnahme. Bei der sich anschließenden internistischen Diagnostik ergab die sonographische Untersuchung des Abdomens und Retroperitoneums als Zufallsbefund eine ca. 5 x 6 cm große, unregelmäßig begrenzte Raumforderung der linken Niere mit atypischer Struktur, die neben liquiden auch sehr echoreiche Veränderungen aufwies.
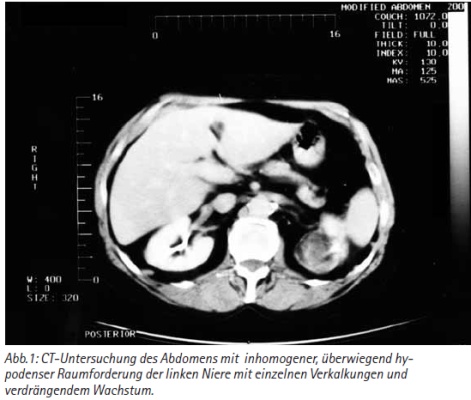
Durch eine CT-Untersuchung der Nieren wurde der dringende Verdacht auf einen Nierentumor bestätigt. Es fand sich eine sehr inhomogene, überwiegend hypodense Raumforderung mit einzelnen Verkalkungen, die nach Kontrastmittelgabe einen inhomogenen Dichteanstieg zeigte. Die Nierenkontur wurde nur gering im mittleren Drittel an der Konvexität der Niere überragt. Das linke Nierenbecken war jedoch nach dorsal verlagert. Eine Harnstauung bestand nicht. Eine Kontinuitätsüberschreitung der linken Niere ließ sich nicht nachweisen. Für einen Befall von Lymphknoten lag kein Anhalt vor und auch in den Oberbauchorganen waren keine Metastasen nachzuweisen [Abb. 1]. Unter dem dringenden Verdacht eines intrarenal gelegenen, lokal begrenzten Nierentumors, ohne Hinweis auf Lymphknoten- und Fernmetastasen, jedoch mit verdrängendem Wachstum gegen das Nierenbecken, erfolgte der Entschluss zur Nierenfreilegung und Nephrektomie. Intraoperativ konnte ein Zystengrundkarzinom nicht ausgeschlossen werden. Makroskopisch bestand am oberen Nierenpol eine zystische Veränderung mit blaßgrauem und bröckligem Zysteninhalt.
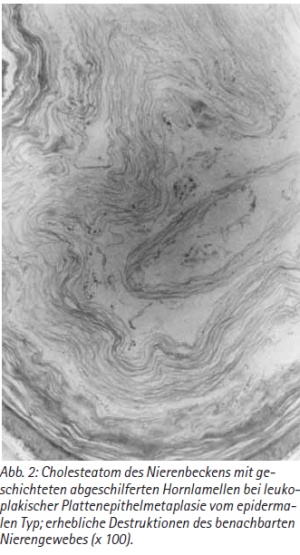
Das umgebende Nierenparenchym war erheblich geschrumpft, das Nierenbecken hydronephrotisch erweitert. Histologisch bestand ein sogenanntes Cholesteatom des Nierenbeckens mit geschichteten abgeschilferten Hornlamellen bei leukoplakischer Plattenepithelmetaplasie vom epithelialen Typ. Daneben fielen erhebliche Destruktionen des benachbarten Nierenparenchyms mit atrophischen Tubuli sowie zahlreichen hyalinisierten und vernarbten Glomeruli auf [Abb. 2]. Des weiteren zeigten sich einzelne Granulome im Bereich der Nierenpapillen, die am ehesten Uratablagerungen entsprachen. Die Markkegel erschienen mit deutlicher interstitieller Fibrose. Außerdem ließ sich eine mäßige unspezifische chronische Pyelitis und eine geringe Hydronephrose diagnostizieren. Schnitte des proximalen Ureters zeigten ein regelmäßiges Urothel.
Der postoperative Verlauf gestaltete sich komplikationslos. Die Entlassung der Patientin erfolgte am 10. postoperativen Tag mit primär heilender Wunde und bei subjektivem Wohlbefinden.
Diskussion
Das sogenannte Cholesteatom der Niere ist eine äußerst seltene tumorähnliche Läsion der ableitenden Harnwege, auf der Grundlage einer Plattenepithelmetaplasie mit einer ausgeprägten Verhornungstendenz [1, 2]. Diese Entität stellt in der Praxis eine Herausforderung für die klinische und radiologische Differentialdiagnostik dar, mit entsprechend weitreichender Konsequenz für die Therapie und Prognose der Patienten. Die klinische Symptomatik zeigt nur wenige eindrückliche, typische Krankheitszeichen auf, so dass die Patienten oft erst spät, bei großem palpablem Tumor aufmerksam werden bzw. der Tumor als Zufallsbefund diagnostiziert wird. Der mehrfache Hinweis von Hornlamellen und – schüppchen im Harnsediment kann den begründeten Verdacht auf ein Cholesteatom erhärten [1, 3]. Nur selten treten jedoch Nierenkoliken beim Abgang von Hornlamellen auf, die eine differentialdiagnostische Abgrenzung zu Koliken anderer Ursache erfordern [3]. Die Diagnostik des Cholesteatoms beruht vorwiegend auf der Sonographie und Computertomographie, besonders wenn es sich um ausgedehntere Befunde handelt. Bei kleineren leukoplakischen Herden können bei der Kontrastmittelapplikation im Ausscheidungsurogramm bzw. retrograden Ureteropyelogramm auffallende Füllungsdefekte hilfreich sein.
Das im vorliegenden Fall beschriebene Cholesteatom trat linksseitig auf. Die Bevorzugung der linken Niere scheint ein häufiger beobachtetes Phänomen zu sein, wovon in der Literatur wiederholt und mit auffallender Übereinstimmung berichtet wird [3, 4, 5]. Primäre retroperitoneale Tumoren wie das Cholesteatom, die ihren Ausgang nicht von den im Retroperitonealraum gelegenen parenchymatösen Organen nehmen, sind eher selten anzutreffen [4]. Cholesteatome des Nierenbeckenkelchsystems auf der Basis einer vorhandenen Plattenepithelmetaplasie werden in der gesamten Weltliteratur bisher mit nur ca. 80 Fällen angegeben [3, 6]. Diese seltene Inzidenz bedingt zweifellos die bis heute differenten Ansichten über das histologische Verhalten des Cholesteatoms, den Verlauf und die Therapie. Hinzu kommt die Tatsache, dass unterschiedliche pathomorphologische Begriffe für die Beschreibung dieser Entität verwendet werden. So wird das Cholesteatom der Niere auch als Leukoplakie der ableitenden Harnwege [2] oder als verhornende desquamative Plattenepithelmetaplasie bezeichnet [6]. Letzterer scheint sowohl dem histologischen Erscheinungsbild als auch dem Krankheitsverlauf am exaktesten Rechnung zu tragen. Dennoch hat die Bezeichnung Cholesteatom als deskriptiver Begriff seine Berechtigung.
Die Leukoplakie hingegen impliziert eine obligate Präkanzerose. Während einige Autoren demzufolge von einem gutartigen Prozess ausgehen und selbst bei langen Beobachtungszeiträumen keine maligne Entartung beobachteten [3, 6], schließen andere die Möglichkeit der Karzinomentstehung mit ein [2]. Ungeachtet dessen ist eine gewisse Rezidivrate unbestritten, so dass die Nephrektomie der Cholesteatom-tragenden Niere bislang als klassische Therapie galt [6]. Hier beginnt sich jedoch mit der Einführung organerhaltender OP-Verfahren ein Wandel abzuzeichnen. So wurde von Kremer und Mitarbeitern (1992) über die erfolgreiche ureterorenoskopische Abtragung kleiner metaplastischer Urothelareale im Nierenbeckenkelchsystem berichtet [3]. Ebenso wurde die Entfernung über eine perkutane Pyeloskopie wiederholt durchgeführt [5]. Allerdings handelte es sich in diesen Fällen um minimale, günstig lokalisierte Befunde, die eine Größenausdehnung von wenigen Millimetern nicht überschritten.
Eine völlige und restlose Beseitigung der metaplastischen Matrix ist bei diesen Verfahren jedoch sehr unwahrscheinlich, so dass unter Umständen mit Rezidiven gerechnet werden muss, die eine Wiederholung der operativen Maßnahmen erforderlich machen können. Für sehr kleine Plattenepithelmetaplasien, die unter Verwendung des intraoperativen Schnellschnittes als sicher benigne abgegrenzt werden können, bieten sich die genannten organerhaltenen Verfahren zweifellos als Methode der Wahl an. Handelt es sich jedoch um große ausgedehnte Cholesteatombefunde mit ungünstiger Lokalisation und lässt sich das Vorliegen eines weit häufigeren malignen Nierentumors nicht sicher ausschließen, lässt sich ein radikales operatives Vorgehen nicht vermeiden, womit aber gleichzeitig die Gefahr einer Rezidivneigung ausgeschlossen wird.
Fallbericht 2
Primäres Osteosarkom der Niere
Eine 49-jährige Patientin fiel im Rahmen einer urologischen Routinesonographie wegen bekannter Nierenzysten mit einem großen soliden linksseitigen Nierentumor auf. Klinisch war die Patientin bis dahin symptomlos. Die Tumordiagnose einer 11 x 8 cm großen Raumforderung mit dem dringenden Verdacht auf ein Nierenzellkarzinom wurde durch ein CT gesichert [Abb. 3]. Die Staging- Untersuchungen (CT, Skelettszintigraphie) ergaben keinen Anhalt für eine Lymphknotenoder Fernmetastasierung. Paraklinisch fielen eine Leukozytose von 11,9x109 /l, eine CRPErhöhung auf 57,8 mg/l, ein LDH von 785 U/l sowie eine Erhöhung der alkalischen Phosphatase von 364 U/l auf. Es wurde eine retroperitoneale Tumornephrektomie durchgeführt, wobei intraoperativ ein „steinharter“ Tumor imponierte.
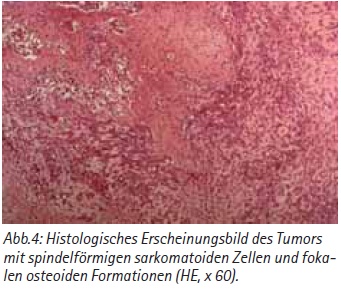
Im Gegensatz zur CT-Untersuchung fanden sich jedoch ausgeprägte paraaortale Lymphknotenpakete. Histologisch wurde ein primäres Osteosarkom der Niere mit typischem spindelzelligem, sarkomatoidem Bild und Osteoidformationen diagnostiziert [Abb. 4]. Die Immunhistochemie zeigt eine positive Reaktion für Vimentin. Die Patientin entwickelte im weiteren Verlauf 4 Wochen nach der Primäroperation ein großes Lokalrezidiv, das einer chirurgischen Therapie nicht mehr zugänglich war [Abb. 3]. Ein Chemotherapieversuch mit Adriamycin verlief frustran. Die Patientin verstarb in massivem Progress 2 Monate nach der Diagnosestellung.
Diskussion
Primäre Osteosarkome der Niere sind extrem seltene Tumorläsionen. Die Erstbeschreibung erfolgte 1936 bei einer Autopsie durch Haining und Poole [7]. Seither sind im internationalen Schrifttum nur 27 Kasuistiken berichtet worden. Extraskelettale Osteosarkome umfassen etwa 4 % aller osteogenen Sarkome und entwickeln sich in parenchymatösen Organen (Lunge, Niere) doppelt so häufig wie in extraskelettalem Weichgewebe [8].
Obwohl primäre renale Sarkome ca. 1 % aller primären renalen Malignome repräsentieren, treten Osteosarkome der Niere extrem selten auf [9, 10].
Die Erklärung der Histogenese stützt sich auf die bislang nicht widerlegte, von Virchow 1884 postulierte Metaplasietheorie [11]. Darin wird die Entstehung dieser Tumorentitität durch Reversion von Bindegewebe zu embryonalem Mesenchym mit nachfolgender Differenzierung zu Osteoklasten beschrieben. Diese Theorie ist bis heute nicht widerlegt und wird von vielen Autoren anerkannt [10, 12, 13, 14].
Das Haupterkrankungsalter liegt zwischen der 4. und 8. Lebensdekade. Die Klinik umfaßt häufig Symptome wie Flankenschmerz, palpablen Flankentumor und Makrohämaturie; sie kann jedoch auch völlig symptomlos verlaufen wie im vorliegenden Fall. Die Paraklinik ist nicht spezifisch. Die alkalische Phosphatase scheint kein verlässlicher Marker zu sein, da nur einige wenige Fälle wie der hier berichtete erhöhte Werte zeigen [13].
Als pathognomonisch werden in mehr als 80 % röntgenologisch nachweisbare Kalzifikationen im Tumor, ein sogenanntes „sunburst- like pattern“, beschrieben [8, 9, 15, 16]. Renale Osteosarkome weisen eine ausgeprägte Tendenz zu Lokalrezidiven auf. Differntialdiagnostisch sollten sarkomatoide Nierenzellkarzinome, adulte Wilms-Tumore und Osteosarkommetastasen in Betracht gezogen werden [10]. Immunhistochemische Untersuchungen mit Vimentinpositivität und negativer epithelialer Markerreaktion sind bei der Diagnosesicherung hilfreich, wie im berichteten Fall beobachtet. Therapeutisch ist die operative Entfernung des Tumors, wenn möglich, anzustreben. Die Möglichkeit der Adriamycin- bzw. Doxorubicin-basierten Chemotherapie wird berichtet, jedoch oft mit inadäquatem Ansprechen [14, 15, 17]. Unsere eigenen Erfahrungen mit einem Adriamycin- Schema korrespondieren mit diesen Aussagen der Literatur. Eine initiale Chemotherapie, gefolgt von einer radikalen Nephrektomie würde wahrscheinlich einen größeren therapeutischen Benefit haben [9]. Es bleibt jedoch das Problem der präoperativen Diagnosestellung, so daß in den meisten Fällen eine radikale Nephrektomie mit nachfolgender adjuvanter Chemotherapie in Betracht zu ziehen ist.
Primäre Ostseosarkome der Niere zeigen ein rasantes Wachstum mit deutlicher Tendenz zu Lokalrezidiven. Die Gesamtprognose ist äußerst schlecht und mit einer hohen Letalität bei einer mittleren Überlebenszeit von ca. 7 Monaten verbunden [10, 14].
Fallbericht 3
Asymptomatisches Paragangliom der Harnblase
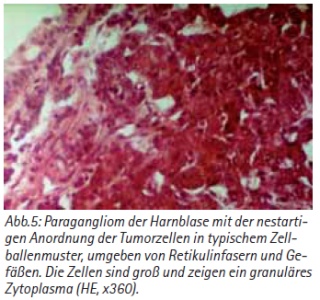
Bei einer 45-jährigen Patientin wurde im Rahmen einer gynäkologischen Routineuntersuchung als sonographischer Zufallsbefund eine Raumforderung an der linken Blasenseitenwand und am Blasendach festgestellt. Zystoskopisch wurde ein solider, intraluminal wachsender Tumor bestätigt, der anamnestisch keine systemischen oder Miktionsbeschwerden verursachte. Nach transurethraler Resektion der Blase (TUR-B) und Nachresektion wurde histologisch die Diagnose eines extraadrenalen Paraganglioms mit infiltrativem Wachstum in die Tunica muscularis gesichert [Abb. 5]. Immunhistochemisch fanden sich positive Reaktionen für die neuronenspezifische Enolase (NSE), Synaptophysin, Chromogranin und Vimentin [Abb. 6]. Eine transurethrale R0-Resektion war nicht möglich. Die radiologische Stagingdiagnostik (Rö-Thorax, CT-Abdomen/Becken) ergab keinen Anhalt für weitere Herde bzw. eine Metastasierung. Paraklinisch waren die Katecholamine im Serum nur grenzwertig erhöht, der Vanillinmandelsäure-Test (VMA) im Urin war negativ. Als definitive Therapie erfolgte eine Blasenteilresektion, wobei ein noch bestehendes großes Residuum des vorresezierten Paraganglioms vollständig entfernt wurde. Die intra- und postoperativen Verläufe waren komplikationslos mit konstant stabilen Kreislaufverhältnissen. Ein Follow-up über 4 Jahre zeigte keinen Rezidivanhalt.
Diskussion
Paragangliome sind eine seltene Erkrankung, die nur 0,06 bis 0,1 % aller Neoplasien der Harnblase ausmachen. Sezernierende Paragangliome bzw. Phäochromozytome sind zu 80 % in den Nebennieren lokalisiert. Ca. 10- 36 % der Paragangliome kommen extraadrenal in verschiedenen Organlokalisationen vor, davon wiederum nur 10 % in der Harnblase [18].
Mehr als die Hälfte der Patienten mit Paragangliomen der Blase bieten eine Symptomatologie mit Hämaturie, hypertensiven Krisen, Tachykardien und Schweißausbrüchen in Verbindung mit Miktionsbeschwerden. Die bevorzugte Tumorlokalisation dieser Tumorläsionen sind die Blasenseitenwände sowie das Blasendach. Frauen sind 3x häufiger betroffen als Männer, insbesondere jüngere Frauen mit einem mittleren Lebensalter von 45 Jahren [19]. Die jüngste bislang berichtete Patientin war ein 10 Jahre altes Mädchen [20]. 10-15 % aller Paragangliome der Harnblase sind maligne und 83 % sind hormonaktiv [21]. Bei hormonaktiven Paragangliomen tritt dabei eine typische Symptomatologie mit Hypertension in 75 %, Hämaturie in 55 % sowie Synkopen, Tachykardien, Kopfschmerzen und Hyperhidrose während und nach der Miktion auf [22]. Eine elektive präoperative adrenerge Blockade ist daher indiziert, um intraoperative hypertensive Komplikationen zu vermeiden [23, 24, 25, 26]. Die präoperative Diagnose gelingt jedoch nur in 40 % der Fälle [27].
Die Diagnostik umfasst neben der Sonographie und der Endoskopie die Katecholaminund Metabolitbestimmung im Serum und 24- Stunden Sammelurin. Selten gibt es Fälle mit normotensiven Blutdrücken trotz hoher Katecholaminkonzentrationen [28].
Andererseits schließen normale Serumkatecholaminwerte Tumore mit sekretorischer Aktivität nicht aus [29]. Im vorliegend berichteten Fall ohne klinische Symptomatik periund intraoperativ war bei nur leicht erhöhten Serumkatecholaminwerten ein negativer VMA-Test im Urin festzustellen. Die Diagnosesicherung erfolgt histologisch und immunhistochemisch mit positiver Reaktion für panendokrine Marker (NSE, Synaptophysin, Chromogranin) [30].
Das Staging wird komplettiert durch CT, MRT und 131 I-MIBG-Szintigrafie (Iodine-131- meta-iodobenzylguanidine) [26, 27, 31].
Die Therapie der Wahl ist die Blasenteilresektion mit R0-Resektion des Tumors. Bei Rezidiven, Malignität bzw. ausgedehntem Tumor kann eine Zystektomie erforderlich sein. Beim Vorliegen von Lymphknotenmetastasen ist eine Lymphadenektomie indiziert. [32]. Spätrezidive und Spätmetastasierungen sind möglich, was in einer entsprechend engmaschigen Nachsorge Berücksichtigung finden muss [33].
Fazit für die Praxis
Die beschriebenen seltenen Tumoridentitäten sind im klinischen Alltag ungewöhnliche Raritäten, die oft nur als Zufallsbefunde diagnostiziert werden. Sie stellen aufgrund ihrer geringen Inzidenz eine differntialdiagnostische Herausforderung dar, die oft eine interdisziplinäre Abklärung erfordern, wie in den vorstehend geschilderten Fällen unter Einbeziehung der modernen Radiologie, Histopathologie und Endokrinologie.
Bei richtiger Diagnosestellung kann in der therapeutischen Konsequenz bei benignen oder semimalignen Tumorläsionen wie dem Cholesteatom oder Paragangliom, wenn möglich, ein organerhaltendes Operationsverfahren angestrebt werden.
Aufgrund der Seltenheit dieser Tumore und der eingeschränkten Erfahrungen, die meistens nur auf Kasuistiken zurückzuführen sind, ist bei nicht sicher auszuschließenden Spätrezidiven eine engmaschige und lebenslange Nachsorge der Patientin zu empfehlen.
Literatur bei den Verfassern.


Datum: 26.05.2011
Quelle: Wehrmedizin und Wehrpharmazie 2011/1