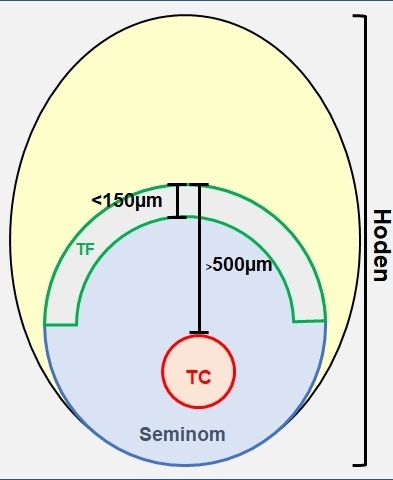
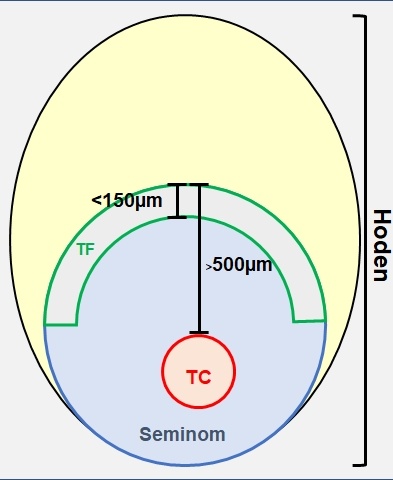
Abb. 1: Schema der lasermikrodissezierten
Areale: Grün die invasive Tumorfront, von der die ersten 150 μm des Tumors
disseziert wurden; Rot das
mindestens 500 μm von der
Tumorfront entfernte zentrale
Tumorkompartiment
¹Bundewehrzentralkrankenhaus Koblenz
²Bundeswehrkrankenhaus Ulm
³Universitätsklinik Köln
Der Hodentumor ist der häufigste Tumor des jungen Mannes und damit auch bei Soldaten, weshalb er wehrmedizinisch relevant ist [1]. Durch die Bundeswehrkrankenhäuser (BwKrhs´er) werden aktuell rund 10 % aller Hodentumorpatienten pro Jahr in Deutschland behandelt.
In Untersuchungen an Kolorektal- oder Bronchialkarzinomen wurden in der invasiven Tumorfront im Vergleich zu vitalen zentralen Tumorarealen unterschiedlich exprimierte Gene gefunden, die mit Prozessen der Metastasierung assoziiert sind [5, 6].
Daher war das Ziel der Studie, einen tieferen Einblick in die Tumorbiologie von Hodentumoren zu erhalten und unterschiedlich exprimierte Gene in der Tumorinvasionsfront im Vergleich zum Tumorzentrum zu identifizieren, die wichtig für Invasion, Progression und schließlich für die Metastasierung sind. Dieses bildet die Grundlage für eine spezifischere Diagnostik und vor allem für eine patientenindividualisierte Therapie metastasierter Hodentumorpatienten. Ideal wäre zudem eine Gensignatur im Primärtumor zu identifizieren, die den Metastasierungsstatus vorhersagen könnte.
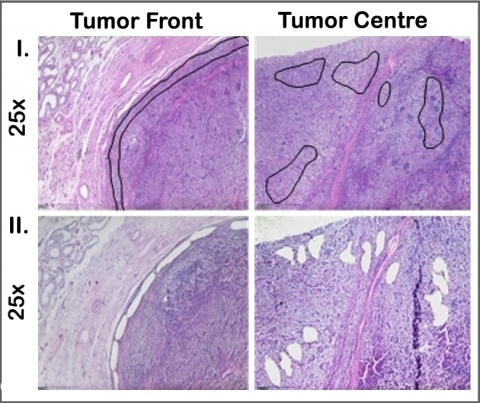
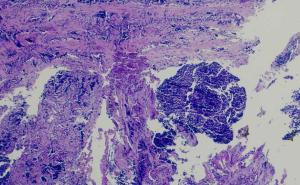
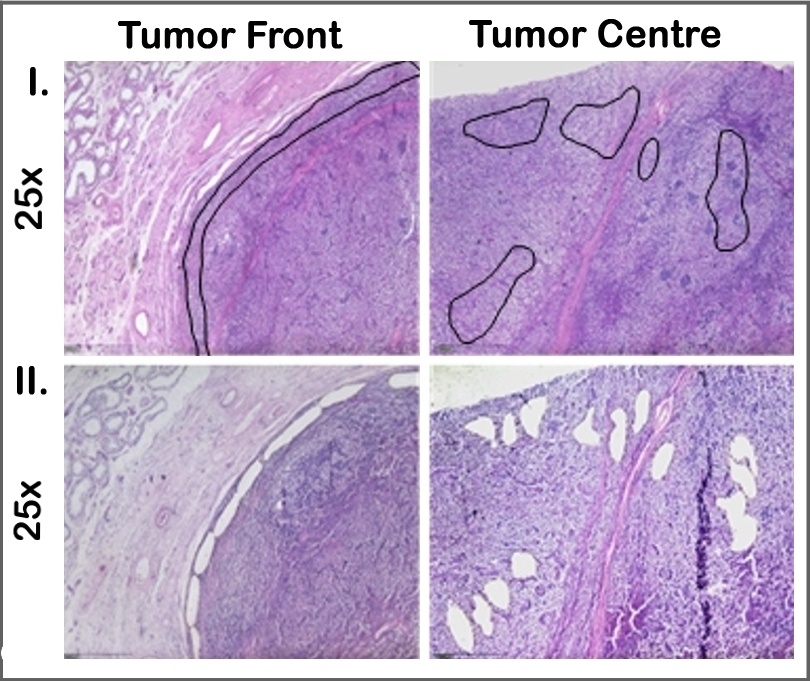
Zunächst wurden alle HE-Schnitte gescreent und die Areale von Tumorinvasionsfront und Tumorzentrum identifiziert. Die weiteren Untersuchungen erfolgten an Formalin-fixiertem (FFPE) Gewebe. Als Tumorinvasionsfront wurde der Tumorbereich definiert, der in den ersten 150 µm in das gesunde Hodengewebe infiltrierend wächst. Das zentrale Tumorareal ist mindestens 500 µm von der Invasionsfront entfernt und berücksichtigt ausschließlich vitale Bereiche. Diese Areale wurden bei jedem Patienten mittels Lasermikroskop disseziert (Abbildungen 1 und 2). Hieraus wurde die RNA nach Standardprotokoll isoliert und 740 bekannte und klinisch relevante Onkogene bzw. Tumorsuppressorgene mittels NanoString PanCancer Progression Panel (NanoString Technologies, WA, Seattle, USA) analysiert. Hier wurde die nCounter® Technologie verwendet, die auch an stark fragmentierter RNA aus FFPE-Gewebe zuverlässig und reproduzierbar anwendbar ist.
Die differenzielle Genexpression wurde als Verhältnis der Expression von Tumorinvasionsfront zu Tumorzentrum für jedes Gen als log2 fold change berechnet und der adjustierte p-Wert angegeben, wobei p<0,05 als signifikant gewertet wurde. Zusätzlich wurden mittels Lasso-Regression (maschinelles Lernen) und Kreuzvalidierung Gensignaturen für Tumorinvasionsfront und Tumorzentrum getestet, die mit einer Auswahl möglichst weniger Gene die Metastasierung vorhersagen soll.
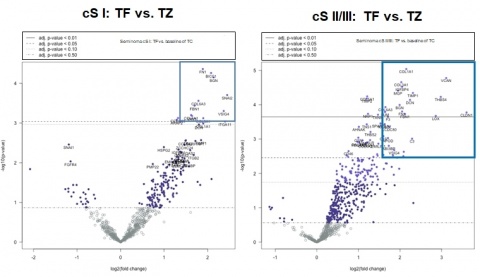
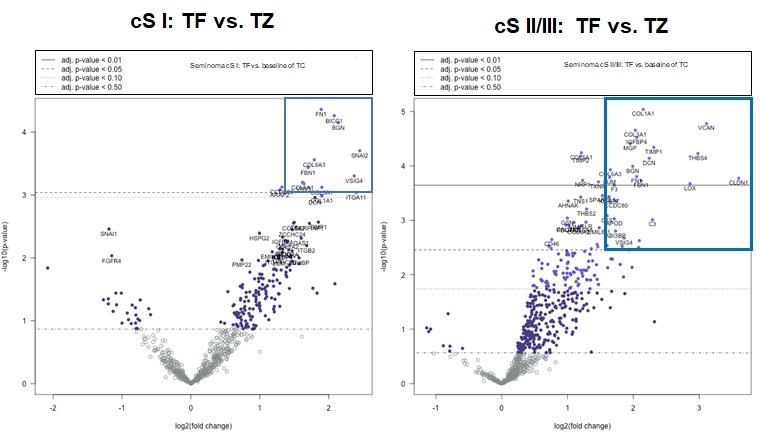
Die hierarchische Clusteranalyse ergab keine eindeutige Aufteilung in metastasierte und nicht metastasierte Patienten. Im Vergleich zwischen Tumorinvasionsfront und Tumorzentrum waren in der nichtmetastasierten Kohorte 12 Gene mit einem log2 fold change >1,5 signifikant unterschiedlich exprimiert und in der Gruppe der metastasierten Patienten 30 Gene (Abbildung 3). Erste Pathway-Analysen zeigen signifikante Expressionsunterschiede zwischen beiden Gruppen u. a. in dem Cluster der Prozesse mit Bezug zum Immunsystem. Die Analyse von vermehrt- bzw. vermindert exprimierten Genen im Vergleich zwischen Tumorinvasionsfront und Tumorzentrum zeigte eine hohe Variabilität zwischen den individuellen Patienten.
Die Lasso-Regression für die Subgruppe der Tumorinvasionsfront ergab eine Gensignatur aus sechs Genen (EPS8L1, ERBB3, FIGF, SLC2A1, SLPI, VAV2), die nahezu perfekt zwischen metastasierten und nicht-metastasierten Patienten unterscheiden kann (Spezifität 100 %, Sensitivität 92,9 %). In der Subgruppe des Tumorzentrums wurde jedoch keine Gensignatur gefunden.
Diese Arbeit zeigt erstmalig, dass seminomatöse Hodentumore, die bisher histopathologisch als uniforme Tumore beschrieben wurden, intertumoral heterogen sind und unterschiedlich exprimierte Gene zwischen metastasierten und nicht-metastasierten Patienten aufweisen. Zusätzlich konnten wir auch intratumoral – anhand einer Vielzahl unterschiedlich exprimierter Gene zwischen den Arealen der Tumorinvasionsfront und des Tumorzentrums – unterschiedliche Expressionsmuster detektieren. Diese Kenntnis ist tumorbiologisch von besonderer Bedeutung und könnte eine mögliche Erklärung für frühe Rezidive unter Surveillance oder nach Chemotherapie metastasierter Seminome darstellen. In einer Folgestudie werden wir unterschiedlich exprimierte Gene von Patienten mit Rezidiv unter Surveillance untersuchen und solche von Patienten mit Rezidiv nach Chemotherapie.
Ebenfalls ist bemerkenswert, dass anhand von sechs Genen, welche in der Tumorinvasionsfront unterschiedlich exprimiert sind, eine Metastasierung vorhergesagt werden kann. Für das Tumorzentrum ist dies jedoch auch unter Berücksichtigung aller 740 Gene nicht möglich. Basierend auf diesen Genen könnte ein neuer und spezifischer Ansatz etabliert werden, der in der Primärdiagnostik eine sichere Unterscheidung ermöglicht und zu einer individualisierten Therapie führt. Dies werden wir an einem unabhängigen multiinstitutionellen Kollektiv, möglichst prospektiv, validieren. Die signifikant unterschiedlich exprimierten Gene werden wir weiter untersuchen – mit Blick auf mögliche Angriffspunkte für eine individualisierte und zielgerichtete Therapie.
Literatur
Oberstabsarzt Dr. Tim Nestler
E-Mail: [email protected]
Datum: 08.02.2019

In Feldkirchen zeigt der Sanitätsdienst seine Leistungsfähigkeit bei der medizinischen Versorgung von Verwundeten. Bei der Informations- und Lehrübung des Sanitätsdienstes der Bundeswehr 2025…

Die Erkenntnis, dass sich Deutschland auf die Notwendigkeit einer Landesverteidigung im Rahmen der Bündnisverteidigung einzustellen hat, ist inzwischen in Mitte der Gesellschaft angekommen.…
Lungenkrebs ist weltweit nach wie vor die häufigste krebsbedingte Todesursache und geht mit hoher krankheitsbedingter Morbidität einher. Aufgrund der uneinheitlichen und oft spät auftretenden…

Die Pariser Special Operation Forces (SOF) Combat Medical Care (CMC) Conference ging am 15. und 16. Oktober 2024 in ihre zweite Runde. Im Jahr 2022 erstmalig als Satellitenkonferenz der CMC in…

Die gezielte Entwicklung von körperlicher Leistungsfähigkeit und Belastungsresilienz ist essenziell für den Aufbau kriegsfähiger Streitkräfte. Besonders relevant erscheint dahingehend die…

Taktische Medizin und taktische Verwundetenversorgung sind ein zentrales Teilgebiet der modernen Notfallmedizin, das sich aus militärischen Erfahrungen entwickelt hat und heute in zahlreichen…

Irregular warfare disrupts conventional medical evacuation, requiring prolonged casualty care with limited resources. Damage control resuscitation, mobile surgical platforms, and prehospital…

Der Krieg in der Ukraine zeigt deutlich, wie zukünftige militärische Auseinandersetzungen sich auf die sanitätsdienstliche Versorgung auswirken können. Aus dem Erfahrungsgewinn lassen sich…

Heute hat der Beauftragte PTBS im Verteidigungsministerium, Oberstarzt Prof. Dr. Peter Zimmermann die Infokampagne "Angehörigenfragebogen PTBS" gestartet.

• Ein Lehrstuhl auf dem Firmengelände – und ein Meilenstein für Forschung, Lehre und Innovation im Schulterschluss mit der Industrie • Das Institut für Medizintechnik und Elektrotechnik…

{kind=link}
{kind=link}
{kind=link}