
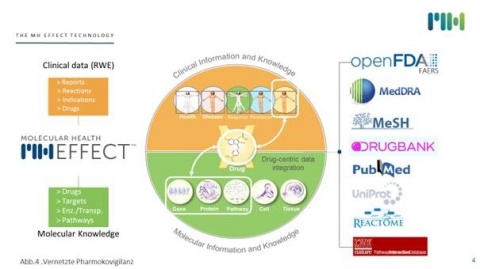
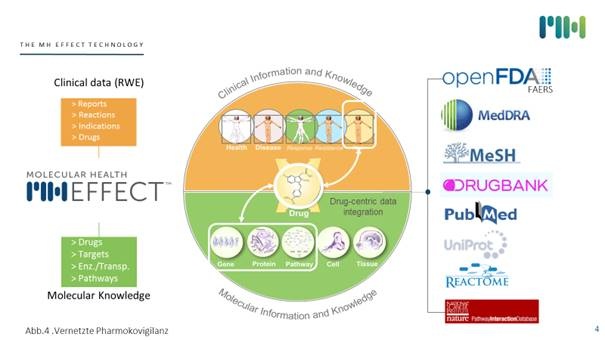
Abb. 3: Molekulare Pharmakovigilanz über die FDA Datenbank FAERS
Unter Arzneimittelsicherheit in der Pharmakologie versteht man die Wissenschaft und Aktivitäten, die zur Entdeckung, Beurteilung sowie zum Verständnis und zur Vorbeugung von unerwünschten Wirkungen oder anderen Problemen in Verbindung mit Arzneimitteln dienen (WHO 2002). Oberstes Ziel der Pharmakovigilanz ist und bleibt es, Menschen vor Schaden durch unerwünschte Wirkungen von Arzneimitteln zu bewahren und den sicheren und effektiven Gebrauch von Arzneimitteln zu fördern.
Pharmakovigilanz ist eine multidisziplinäre Kooperation von Ärzten, Apothekern und Naturwissenschaftlern, die in aller Konsequenz dem Schutz der Patienten vor Risiken und Nebenwirkungen von Arzneimitteln dient. In der Gebrauchsinformation beschreibt das jeweilige Pharma-Unternehmen den zugelassenen Anwendungsbereich des Arzneimittels und informiert über relevante Risiken sowie über Nebenwirkungen. Bekannt ist, dass nicht jede Nebenwirkung bei jedem Patienten auftreten muss. Nebenwirkungen müssen stets in Relation zum Schweregrad der Erkrankung und zum Nutzen des Arzneimittels bzw. den Bedarf für den jeweiligen Patienten gesehen werden. Sie sind teilweise schädliche und unbeabsichtigte Reaktionen auf das Arzneimittel. Diese können beim bestimmungsgemäßen Gebrauch, aber auch von Fehl-, Miss- bzw. Off-Label-gebrauch (bewusste Anwendung außerhalb der Zulassung) und Medikationsfehler (unbeabsichtigter Fehler im Medikationsprozess), dem Patienten zu schaden. Die Therapietreue (Compliance) des Patienten d. h. die Einhaltung des vom Arztes vorgegebenen Therapieschemas spielt eine entscheidende Rolle innerhalb des Medikationsprozesses und sollte daher auch bei der Erfassung und Bewertung von Nebenwirkungen berücksichtigt werden. Insbesondere im Zuge von Langzeittherapien oder Erkrankungen, die zunächst ohne größere Beschwerden verlaufen können, ist die Therapietreue häufig unzureichend. Durch sachgerechte Aufklärung und Beratung der Patienten durch Arzt und Apotheker sollten Zweifel des Patienten ausgeräumt und Nebenwirkungen im Kontext des Nutzens für den Patienten aufgezeigt werden. Dies kann die Therapietreue des Patienten erhöhen. Grundsätzlich sollten der Therapiebedarf des Patienten sowie die Folgen einer unterlassenen Therapie berücksichtigt werden.
Unter einer Arzneimittelwechselwirkung (Arzneimittelinteraktion) wird eine Veränderung des Effektes durch die gleichzeitige Gabe eines anderen Arzneimittels verstanden. Derartige Arzneimittelwechselwirkungen sind in vielen Fällen erwünscht und erhöhen die Effektivität von Kombinationstherapien wie z. B. Kombination von Antihypertonika oder die Kombination von Analgetika. Ebenso basiert die pharmakologische Behandlung einer Intoxikation auf dem Prinzip der Arzneimittelinteraktion bzw. Arzneimittel-Toxin-Interaktion wie z. B. Naloxon bei Opinoidvergiftung oder Physostigmin bei Atropin Überdosierung. Auch die Ernährung kann die Wirkung von Arzneimitteln stark verändern. Die Folgen können erheblich, wenn nicht sogar tödlich sein. Nehmen betagte Patienten zu viele Medikamente gleichzeitig ein, steigt durch diese Polymedikation die Wahrscheinlichkeit von unerwünschten Wechsel- und Nebenwirkungen.
Aber auch als gesund geltende Lebens- und Nahrungsergänzungsmittel können die Wirkung von Arzneimitteln verändern und das ganz unabhängig vom Alter der Person. Es gibt nicht ohne Grund immer den Hinweis auf Beipackzetteln, ob ein Medikament vor, beim oder nach dem Essen eingenommen werden soll. So muss beispielsweise das Schilddrüsenhormon L-Thyroxin eine halbe Stunde vorher eingenommen werden, sonst interagiert es mit dem Essen und wird nicht richtig freigesetzt.
Um nach der Zulassung eines Arzneimittels dessen Nutzen-Risiko-Verhältnis weiter im Lichte wachsender Erkenntnis zu überprüfen, werden alle relevanten Daten fortlaufend vom pharmazeutischen Unternehmer sowie von den zuständigen Behörden überwacht und bewertet. Die Kenntnisse über die Sicherheit von Arzneimitteln sind zum Zeitpunkt ihrer erstmaligen Zulassung nicht vollständig. Dies ergibt sich vor allem daraus, dass die klinische Erprobung eines Arzneimittels an einer relativ geringen Zahl von Patienten durchgeführt wird. Seltene oder sehr seltene unerwünschte Wirkungen, Wechselwirkungen oder andere Risiken im Zusammenhang mit der Arzneimittelanwendung können in klinischen Prüfungen üblicherweise nicht erkannt werden. Diese Patientinnen und Patienten sind zudem unter verschiedenen Aspekten für die klinische Prüfung besonders ausgewählt worden, was nicht notwendigerweise den Bedingungen bei der breiten Anwendung des Arzneimittels entspricht. Das Arzneimittelgesetz der Bundesrepublik Deutschland sieht deshalb vor, dass nach der Zulassung eines Arzneimittels die Erfahrungen bei seiner Anwendung fortlaufend und systematisch gesammelt und ausgewertet werden. Dies ist eine der Aufgaben des Bundesinstitutes für Arzneimittel und Medizinprodukte (BfArM). Insbesondere schwerwiegende und bisher unbekannte unerwünschte Wirkungen sind für die Gesamtbewertung eines neuen Arzneimittels von großer Bedeutung. Neue Erkenntnisse über die Sicherheit von Arzneimitteln können sich auch noch lange Zeit nach ihrer Zulassung ergeben und hängen von neuen Entwicklungen in der medizinischen Wissenschaft ab.
Wenn die Bewertung von Arzneimittelrisiken ergibt, dass der Zulassungsstatus von Arzneimitteln dem Stand der wissenschaftlichen Erkenntnis angepasst werden muss, koordiniert das BfArM ggf. in Abstimmung mit den zuständigen Gremien der Europäischen Union (EMA), die notwendigen Maßnahmen. Bei diesen Maßnahmen kann es sich zum Beispiel um Einschränkungen der Anwendungsgebiete eines Mittels handeln, unter bestimmten Bedingungen kann aber auch die Zulassung eines Arzneimittels widerrufen werden. Über derartige Maßnahmen informiert das Bundesinstitut für Arznei- und Medizinprodukte Ärzte, Patienten und Interessierte.
Dazu gehören ggf. auch Handlungsempfehlungen für Patienten, Ärzte zur sicheren Anwendung von Arzneimitteln. Diese setzen sich u. a. aus dem Spontanmeldesystem, aus internationalen Literaturrecherchen sowie Ergebnissen aus weiteren präklinischen oder klinischen Studien, patientenorientierten Programmen, Internet und den sozialen Kommunikationsmedien, die einer systematischen Produktbeobachtung entsprechen, zusammen.
Die Datenbanken des Bundesinstitutes für Arznei und Medizinprodukte (AfArM ) sowie die ABDA-Datenbank, Bundesvereinigung der Apothekerverbände, sind die umfangreichsten Datenbanken zu Interaktionen mit Schwerpunkt auf den in Deutschland aktueller und früher eingesetzten Medikamenten. Es sind Wechselwirkungen zwischen Stoffen und Produkten (Fertigarzneimittel, Nahrungsergänzungsmittel, Diätetika, Medizinprodukte) hinterlegt. Die Warnungen enthalten immer Hinweise zum Schweregrad der Interaktion und häufig auch Empfehlungen zum weiteren Vorgehen.
Die unter anderem oben beschriebenen Auswertungen der Pharmakovigilanzdatenbanken erkennen “Arzneimittelinteraktionen“ und an “Interaktionen beteiligte Medikamente“. Die Ergebnisse einer auf der Basis der US-amerikanischen FDA-Pharmakovigilanzdaten durchgeführten Risikoanalyse sind vom WHO Pharmakovigilanzzentrum publizierte Übersichten über die häufigsten Interaktionstypen. Auffällig ist, dass das Gros der Meldungen (86 %) sich auf durch die Interaktion verstärkte Toxizität von Arzneistoffen bezieht. Wirkungsverluste, bedingt durch Interaktionen, wurden wesentlich seltener gemeldet. Tatsächlich fallen toxische Effekte klinisch stärker auf. Wirkungsverluste hingegen sind nur bei einigen besonderen Medikamentengruppen problematisch (z. B. Transplantatabstoßung bei Immunsuppressiva oder Resistenzbildung bei unzureichenden Plasmakonzentrationen und damit unzureichender antimikrobieller Wirkung von Antibiotika. Darüber hinaus treten die indirekten, funktionellen pharmakodynamischen Interaktionen auf, dicht gefolgt von indirekten pharmakokinetischen Interaktionen, v. a. bedingt durch Veränderung von Enzymaktivitäten. In ca. 20 % der beobachteten Interaktionen ist der genaue Mechanismus immer noch unbekannt.
Pharmakogenetische Untersuchungen beschäftigen sich mit dem Einfluss genetischer Eigenschaften auf die Pharmakodynamik (Wirkqualität und -stärke), die Pharmakokinetik (Geschwindigkeit der Aufnahme, Metabolisierung und Ausscheidung) von Arzneistoffen sowie auf das Risiko von Überempfindlichkeitsreaktionen. Die pharmakogenetische Untersuchung berücksichtigt auch die klinische Validität des Arzneimittels und deren klinischen Nutzen. Die Möglichkeiten der molekulargenetischen Diagnostik und die Durchführung molekulargenetischer Analysen zum Nachweis einzelner genetischer Polymorphismen gewinnt zunehmend an Bedeutung, insbesondere bei genetisch bedingter Unverträglichkeit von Medikamenten. Genetische Polymorphismen sind Sequenzvariationen, die, wenn sie in einem funktionellen Bereich des menschlichen Genoms liegen, einen pathogenetischen Einfluss haben können. Das Auftreten einer einzelnen Sequenzvariation kann dazu führen, dass ein Stoffwechselweg eingeschränkt oder vollständig gestört ist. Dabei kann eine akute oder chronische Exposition gegenüber exogenen Noxen zu einem uneinheitlichen Krankheitsbild führen. Genetische Polymorphismen treten mit einer Häufigkeit von 1 bis 50 % in einer Bevölkerung auf. Bereits seit vielen Jahren ist bekannt, dass Polymorphismen bei Einnahme von Arzneimittelwirkstoffen zu genetisch bedingten unerwünschten Arzneimittelreaktionen führen können. Aber auch bei genetisch bedingten Nahrungsmittelunverträglichkeiten oder bei chronischer Exposition gegenüber Schadstoffen hat die Bestimmung genetischer Polymorphismen einen hohen differentialdiagnostischen Nutzen. Da genetische Polymorphismen erblich erworben werden und lebenslang unverändert bleiben, liegt einer der Vorteile der Diagnostik in der einmaligen Durchführung. Die Sequenz eines Gens bzw. die einzelnen Nukleotide lassen sich mit Hilfe molekulargenetischer Verfahren routinemäßig in einem entsprechend spezialisierten Labor untersuchen. Im wissenschaftlichen Mittelpunkt der vergangenen Jahre stand die Lokalisation der Gene auf den menschlichen Chromosomen, deren Sequenzanalyse und inwieweit Sequenzvariationen zu einer Funktionsänderung der Proteine führen. Folge sind Komplikationen im Stoffwechsel endogener (z. B. Fettstoff-, Vitamin-, Hormonstoffwechsel) und exogener Substanzen (z. B. Nahrung, Chemikalien, Arzneimittel). Manche der Sequenzvariationen können für das tägliche Leben wichtig sein, während andere zunächst völlig unauffällig sind. So führt beispielsweise das Vorliegen einer Sequenzvariation im Laktase Gen zu einer Milchzuckerunverträglichkeit (Laktoseintoleranz), während eine genetische Variante, die zu einem veränderten Abbau eines Arzneimittels beiträgt, erst dann auffällig wird, wenn die Einnahme des Medikaments zu einer unerwünschten Arzneimittelwirkung geführt hat. Die Kenntnis über das Vorliegen genetischer Polymorphismen hat in der Folge dazu beigetragen, dass die Zahl molekulargenetischer Untersuchungen mit unterschiedlichen Fragestellungen ständig zugenommen hat. Während bei monogenen Erkrankungen häufig eine vollständige Sequenzanalyse des betroffenen Gens erforderlich ist, reicht bei der Untersuchung von Polymorphismen der punktuelle Nachweis der Sequenzvariation mit moderatem technischem Aufwand. Genetische Polymorphismen sind dadurch definiert, dass sie in einer Population mit einer Häufigkeit von mindestens ein Prozent auftreten. Polymorphismen mit einer Häufigkeit von weniger als ein Prozent werden als Keimbahnmutationen bezeichnet. Keimbahnmutationen betreffen im Gegensatz zu somatischen Mutationen den gesamten Organismus und werden an die Folgegenerationen vererbt, genauso wie genetische Polymorphismen.
Neuroleptika, Antidepressiva und andere Psychopharmaka zahlreiche Psychopharmaka, aber auch Betablocker wie Metoprolol sowie das in der Behandlung des Brustkrebses häufig eingesetzte Antiöstrogen Tamoxifen werden zu einem erheblichen Teil in der Leber durch das Enzym Cytochrom P450 2D6 (CYP2D6) metabolisiert. Das CYP2D6 Gen ist auf dem menschlichen Chromosom 22 (22q13.1) lokalisiert und innerhalb des Gens befindet sich eine Vielzahl von Sequenzvariationen, die zu einer veränderten CYP2D6 Enzymaktivität führen. Diese genetischen Varianten führen beim Menschen zu unterschiedlichen metabolischen (M) Phänotyen, den sog. poor (PM), intermediate (IM), extensive metabolizer (EM)‘ sowie ‚ultrarapid metabolizer (UM)‘. Personen, die unter Einnahme der empfohlenen Dosis eines Arzneimittels, das zu einem maßgeblichen Teil durch das CYP2D6 Enzym metabolisiert wird, an unerwünschten Arzneimittelwirkungen leiden, können Träger einer solchen Sequenzvariation des CYP2D6 Gens sein. Diese werden zu den sog. ‚schlechten Metabolisierern‘ (‚poor metabolizer‘) gezählt. Etwa 5 bis 10 % unserer Bevölkerung tragen diese Sequenzvariationen. Menschen mit einer intermediären CYP2D6 Enzymaktivität (etwa 20 % der Bevölkerung) können diese Arzneimittel in der Regel gut vertragen. Wird allerdings gleichzeitig mit Wirkstoffen therapiert, die ebenfalls durch CYP2D6 Enzyme metabolisiert werden, kann ein Wettbewerb zwischen diesen Wirkstoffen entstehen. Insbesondere, wenn gleichzeitig mit Arzneimitteln therapiert wird, die das Enzym CYP2D6 hemmen, führt die dadurch ausgelöste CYP2D6 Inaktivierung zu einer Störung des Arzneimittelstoffwechsels. Da das CYP2D6 Enzym gehemmt wird treten Betroffene plötzlich als ‚schlechte Metabolisierer‘ (PM) phänotypisch in Erscheinung. Hingegen werden bei Menschen mit hoher CYP2D6 Enzymaktivität (ultrarapid metabolizer, ca. 3 % unserer Bevölkerung) die Wirkstoffe viel zu schnell metabolisiert und die therapeutische Wirkung bleibt aus. Es werden in der Literatur beispielsweise Vorschläge zur Dosisanpassung verschiedener Psychopharmaka gemacht, um eine Therapieoptimierung zu erzielen und dem Auftreten genetisch bedingter Arzneimittelunverträglichkeiten vorzubeugen. Bei einer Langzeittherapie, zu deren Behandlung der Patient möglicherweise jahrelang Medikamente mit entsprechenden Nebenwirkungen einnehmen muss, ist die sichere Diagnose der Erkrankung essentiell. Hier gewinnen Biomarker mehr und mehr an Bedeutung, denn sie können eine schwierige Diagnose absichern oder sie sogar erst ermöglichen. Einer Reihe von Erkrankungen wie zum Beispiel bestimmten Krebserkrankungen, der Alzheimer-Erkrankung oder der rheumatoiden Arthritis geht häufig ein frühes, symptomloses Krankheitsstadium voraus. In dieser Phase helfen Biomarker, symptomfreie Risikopersonen rechtzeitig und zuverlässig zu identifizieren.
MH Effect integriert nicht nur mit Hilfe einer molekularen Schlüssel- und Kerntechnologie Daten aus dem Bereich der Arzneimittelsicherheit sondern validiert sie mit klinischem und molekularem Wissen unter anderem für die Pharmaindustrie und die Aufsichtsbehörden FDA und EMA. Die Bewertung genetischer Eigenschaften hinsichtlich ihrer Bedeutung für die Wirkung eines Arzneimittels bei einer Behandlung ist Aufgabe der biomedizinischen Wissenschaft. Im Gegensatz zur Identifizierung genetischer Ursachen für die Entstehung seltener (monogener Formen von) Erkrankungen ist die Feststellung des genetischen Anteils bei der interindividuellen Variabilität von Arzneimittelwirkungen oftmals schwierig bzw. es sind hierfür nur wenige geeignete Evaluierungsmöglichkeiten vorhanden. Die Effizienz von pharmakologischen Wirkstoffen kann teilweise sehr stark schwanken und ist meist abhängig von nicht-erblichen Faktoren (Alter, Ernährungszustand, Erkrankungen, Komedikation etc.). Für den genetischen Anteil bei unerwünschten Arzneimittelwirkungen gibt es derzeit kaum Schätzungen. Die Einschätzung der klinischen Validität und des klinischen Nutzens pharmakogenetischer Untersuchungen basiert auf der jeweils bestmöglichen medizinischen Evidenz. Die Europäische Arzneimittelbehörde (EMA) hat bereits heute etwa 15 % ihrer bisher zugelassenen Arzneimittel mit Hinweisen auf spezifische, sowohl somatische als auch hereditäre pharmakogenetische Eigenschaften in den Produktinformationen versehen. Im Zeitalter der Globalisierung sind auch die Medizin und die Pharmazie in der digitalen Welt angekommen.
Literatur beim Verfasser
Abb. beim Verfasser
Anschrift des Verfassers:
Generalarzt a. D. Dr. Arno M. Roßlau
Unterm Wald 6
31707 Bad Eilsen-Heeßen
E-Mail: [email protected]
Datum: 05.03.2018
Quelle: Wehrmedizin und Wehrpharmazie 4/2017
Eine 31-jährige Patientin entwickelte nach beidseitiger Tonsillektomie ein zervikales Hautemphysem, das sich beidseits zervikal ausbreitete. Die Computertomografie zeigte zusätzlich ein…

Blut und Blutkomponenten sind eine limitierte, jedoch unverzichtbare Ressource in der medizinischen Versorgung – sowohl im zivilen als auch im militärischen Kontext. NATO-Hochrechnungen für den…

Eine Drohne, die Verwundete transportiert? Was 2021 noch wie Science-Fiction klang, ist heute Realität. Bei der Informationslehrübung Sanitätsdienst 2025 zeigt das Unternehmen Avilus seine dritte…

Methoxyfluran ist ein sicheres, schnell wirkendes und effektives Arzneimittel („proof-of-concept“) zur patientengesteuerten Analgesie bei akuten traumaassoziierten, mittelstarken bis starken…

Heute hat der Beauftragte PTBS im Verteidigungsministerium, Oberstarzt Prof. Dr. Peter Zimmermann die Infokampagne "Angehörigenfragebogen PTBS" gestartet.
Lungenkrebs ist weltweit nach wie vor die häufigste krebsbedingte Todesursache und geht mit hoher krankheitsbedingter Morbidität einher. Aufgrund der uneinheitlichen und oft spät auftretenden…

Irregular warfare disrupts conventional medical evacuation, requiring prolonged casualty care with limited resources. Damage control resuscitation, mobile surgical platforms, and prehospital…

Die Pariser Special Operation Forces (SOF) Combat Medical Care (CMC) Conference ging am 15. und 16. Oktober 2024 in ihre zweite Runde. Im Jahr 2022 erstmalig als Satellitenkonferenz der CMC in…

• Ein Lehrstuhl auf dem Firmengelände – und ein Meilenstein für Forschung, Lehre und Innovation im Schulterschluss mit der Industrie • Das Institut für Medizintechnik und Elektrotechnik…

Taktische Medizin und taktische Verwundetenversorgung sind ein zentrales Teilgebiet der modernen Notfallmedizin, das sich aus militärischen Erfahrungen entwickelt hat und heute in zahlreichen…
{kind=link}
{kind=link}