Anwendungsorientierte Forschung ermöglicht die State-of-the-art-Diagnostik von Pockenviren
Aus dem Institut für Mikrobiologie der Bundeswehr, München (Leiter: Oberstarzt Prof. Dr. L. Zöller)
H. Meyer
Zusammenfassung
Hintergrund: Das Variolavirus, der Erreger der Pocken, gilt als das gefährlichste Agens im Rahmen von bioterroristischen Anschlägen. Sollten Pockenerreger freigesetzt werden, führt das zu einer weltweiten Gesundheitskatastrophe. Für Maßnahmen zur Eindämmung ist deshalb eine möglichst rasche Identifizierung des Erregers unerlässlich.
Methoden: Eine spezifische Pocken-Diagnostik wurde auf Basis der Real-time-PCR entwickelt. Die Durchführung von umfangreichen Validierungsarbeiten hatte zum Ziel, die diagnostischen Verfahren nach DIN EN ISO 15189 zu akkreditieren.
Ergebnisse: Ein Algorithmus zur Identifizierung und Differenzierung der Orthopockenviren mittels akkreditierter Untersuchungsverfahren ist am Institut für Mikrobiologie der Bundeswehr (InstMikroBioBw) realisiert. Im Rahmen der Validierungsarbeiten zur Spezifität der Nachweisverfahren erwies sich die umfangreiche Erreger-Stammsammlung als besonders geeignet.
Schlussfolgerungen: Bei Pockenverdacht stehen am InstMikroBioBw geeignete Untersuchungsverfahren zur Verfügung, um eine rasche Diagnose zu stellen. Dies ist Voraussetzung, um Maßnahmen zur rechtzeitigen Eindämmung eines Ausbruchs einzuleiten.
Schlüsselworte: Variolavirus, akkreditierte Diagnostik, Pocken, Genomsequenzierung, Orthopockenviren
Keywords: variola virus, smallpox, orthopoxviruses, dia-gnostics, genome sequencing
Bedeutung der Pocken heute
Das Variolavirus ist der Erreger der Pocken (Variola vera, smallpox), einer sehr ansteckenden, hochfieberhaften und akut verlaufenden exanthematischen Virusallgemeinkrankheit. Vor Beginn des Eradikationsprogrammes der WHO traten pro Jahr bis zu 10 Millionen Fälle auf. 1977 konnte in Somalia dann der letzte natürlich aufgetretene Pockenfall diagnostiziert werden; 1980 erklärte die WHO die Pocken für ausgerottet. Heute wird das Variolavirus – entsprechend einer Klassifizierung durch die Centers for Disease Control and Prevention (CDC) – unter den „Class A Bioterrorist Threats“ als der gefährlichste Erreger eingestuft: Die Infektiosität und Stabilität im luftgetragenen Zustand, die relativ hohe Kontagiosität und Letalität, eine fehlende Immunität der Bevölkerung und die Tatsache, dass erst 7 - 21 Tage nach „Ausbringung“ eine Diagnose gestellt werden kann, führen dazu, dass ein Pockenausbruch heute sich zu einem weltweiten „Public Health“-Notfall entwickeln würde [1]. Die Experten sind sich einig, dass die Wahrscheinlichkeit einer gewollten Freisetzung der Pocken sehr gering ist, gleichwohl der Schaden aber immens wäre. Umso wichtiger ist es, möglichst rasch eine eindeutige und zuverlässige Diagnose zu stellen. Ist der Pockenverdacht bestätigt, so hat das eine Reihe von Maßnahmen zur Folge:
• Isolierung der Pockenkranken,
'• Ermittlung und Quarantäne der Kontaktpersonen,
• Durchführung von Impfungen sowie
• Einsatz antiviraler Mittel.
Auch bei einem leistungsfähigen öffentlichen Gesundheitssystem sind dies enorme organisatorische und logistische Herausforderungen. Es ist daher verständlich, dass die Maßnahmen nicht sofort greifen können und dass es zu weiteren Fällen kommen wird. Berücksichtigt man die hohe Mobilität der heutigen Bevölkerung in Verbindung mit der langen Inkubationszeit, so wird ein Ausbruch der Pocken zwangsläufig zu einer weltweiten Gesundheitskatastrophe führen.
Wie rasch sich ein Virus in der heutigen Zeit über den international vernetzten Flugverkehr verbreiten kann, hat die SARS-Epidemie eindrucksvoll bewiesen (SARS=Schweres Akutes Respiratorisches Syndrom): Von November 2002 bis Juli 2003 erkrankten mehr als 8 000 Personen in 30 Ländern, knapp 1 000 verstarben [2]. Die globale Vernetzung hat aber auch ihre guten Seiten. So konnte die schnell anlaufende internationale Zusammenarbeit, insbesondere auf dem Gebiet der Diagnostik, diese Infektionskrankheit eindämmen. Entscheidend dafür ist eine korrekte Diagnose, denn dann können weitere Maßnahmen (Isolierung, Quarantäne) ergriffen werden, die, konsequent angewendet, zur Eindämmung führen. Zu erwähnen ist der hohe Anteil an medizinischem Personal, das während der SARS-Epidemie erkrankte bzw. verstarb. Damit muss auch bei einem Ausbruch der Pocken gerechnet werden.
Diagnostik bei Orthopockenviren
Stellenwert der Diagnostik
Das InstMikroBioBw befasst sich seit Jahren intensiv mit der Identifizierung und Differenzierung der Orthopockenviren. Im Vordergrund stehen dabei die humanpathogenen Spezies Variola-, Affenpocken-, Kuhpocken- und Vacciniavirus. Affenpockenviren verursachen ein von den Menschenpocken (smallpox) klinisch nicht zu unterscheidendes Bild, und auch Kuhpocken- und Vacciniaviren können insbesondere bei Immunsupprimierten eine generalisierte Infektion hervorrufen. Daher ist es von entscheidender Bedeutung, herauszufinden, um welchen Erreger es sich handelt. Im Falle der ausschließlich humanpathogenen Variolaviren mit ihrem hohen Übertragungspotenzial hängen von der richtigen Diagnose wichtige Entscheidungen ab, wie das oben dargestellte Ausrufen eines weltweiten Public Health-Notfalls. Bei einem falsch-negativen Befund (eine Pockeninfektion wird nicht erkannt) kann sich die Seuche weiter ausbreiten, mehr Menschen werden sich infizieren und die Eindämmungsmaßnahmen laufen erst verzögert an. Bei einem falsch-positiven Befund (eine Affen- oder Kuhpockenvirus-Infektion wird fälschlich als eine Infektion mit Variolavirus angesehen) wird ein Pocken-Alarm mit all seinen politischen und internationalen Konsequenzen ausgelöst. Stellt sich das später als Fehlalarm heraus, tragen meldende Labore und die betreffenden Nationen eine schwere Verantwortung. Eine Pockendia-g-nostik muss daher falsch-negative und falsch-positive Befunde unter allen Umständen vermeiden.
Diagnostische Verfahren
Geeignete, CE-markierte und damit offiziell zugelassene Testverfahren stehen allerdings nicht zur Verfügung, denn es gibt dafür keinen Markt. Daher sind die Nationen auf die Entwicklung von In-House-Testen angewiesen. Eine Übersicht solcher Teste wurde im Auftrag der World Health Organization (WHO) von einer Expertenkommission publiziert [3]. Mit dem Aufkommen moderner molekularbiologischer Nachweistechniken hat sich dabei die Verwendung von Real-time-PCR-Verfahren seitdem als Methode der Wahl etabliert. Das InstMikroBioBw hat schon vor Jahren auf der Basis einer Real-time-PCR ein Verfahren zum Nachweis von Orthopockenviren und gleichzeitiger Differenzierung von Variolavirus publiziert [4]. Da mit Ausnahme der beiden WHO-Referenzlabore für Pocken am Center for Disease Control and Prevention (CDC) in den USA und in Koltsovo, Russland, kein Labor mit Variolaviren arbeiten darf, konnten die notwendigen Validierungsarbeiten mit verschiedensten Variolavirus-Stämmen nur mit diesen beiden Partnern durchgeführt werden. Die umfangreichen, vorgeschriebenen Validierungsarbeiten führten auf dem Gebiet der Diagnostik von Pockenviren zur Erstellung eines Algorithmus, der nicht nur die Identifizierung von Variolaviren, sondern auch die Differenzierung von Affen- und Kuhpockenviren sowie von Vacciniaviren beinhaltet. Dies schließt auch eine orientierende Untersuchung auf Variolaviren mit ein. Dazu ist das InstMikroBioBw (neben anderen Laboren in Deutschland) vom Robert Koch Institut (RKI) als Untersuchungsstelle benannt. Kulturelle Verfahren, die die Vermehrung des Erregers zum Ziel haben, dürfen aber nicht durchgeführt werden. Diese und weitere Arbeiten mit lebendem Virus sind nur in den beiden WHO-Referenzlaboren am CDC und in Koltsovo erlaubt.
Akkreditierung des InstMikroBioBw
Mit über 110 Parametern wurde der Zentralbereich Diagnostik des InstMikroBioBw im September 2012 durch die Deutsche Akkreditierungsstelle nach DIN EN ISO 15189 akkreditiert. Die Akkreditierung erfordert im Gegensatz zu einer Zertifizierung nicht nur die Etablierung und Pflege eines Qualitätsmanagementsystems, sondern darüber hinaus auch den Nachweis eines hohen Standards der mikrobiologischen Diagnostik. Damit wird sichergestellt, dass unter Berücksichtigung der geltenden Normen, strukturellen Veränderungen und dem ständigen wissenschaftlichen Fortschritt die Aufgaben und die Anforderungen an den Nachweis von B-Kampfstoffen und anderen gefährlichen Krankheitserregern und Toxinen in höchster Qualität erfüllt werden. Dies verpflichtet auch zur Bewertung von diagnostischen Verfahren, die von anderen Arbeitsgruppen publiziert werden. Basierend auf einer Literaturrecherche (zusammengefasst von [3]) wurden seit 2002 insgesamt zwölf Variolavirus-spezifische Nachweisverfahren publiziert. In Zusammenarbeit mit den beiden WHO-Referenzlaboren für Pocken und dem Robert Koch Institut (RKI) konnten wir durch experimentelle und in silico Untersuchungen zeigen, dass vier dieser Nachweisverfahren mit bestimmten Kuhpockenviren zu falsch-positiven Reaktionen führen; Kuhpockenvirus wird in diesen Testen also fälschlich als Variolavirus diagnostiziert [5]. Offenbar sind bei diesen Kuhpockenviren Sequenzen konserviert, die sich spezifisch auch bei den Variolaviren finden [6]. Das passt gut zu der Theorie, dass sich ausgehend von einem gemeinsamen Vorfahren, sowohl verschiedene Subtypen von Kuhpockenviren als auch die Variolaviren evolutionär entwickelt haben.
Die Orthopockenvirus-Stammsammlung
Für eine Validierung, insbesondere der Spezifität von diagnostischen Verfahren, ist eine umfangreiche, gut charakterisierte Stammsammlung von enormem
Vorteil. Mit Stand 2015 besteht die Sammlung des InstMikroBioBw aus 279 Stämmen verschiedener Orthopockenviren (siehe Tabelle 1); in Bezug auf Kuhpockenviren besitzt das Institut die weltweit größte Stammsammlung. Die Sammlung besteht aus 88 Affenpockenviren (diese gehören der Risikogruppe 3[1] an), 107 Kuhpockenviren, 22 Vacciniaviren, 50 Kamelpockenviren und 12 Mäusepockenviren. Alle Stämme sind in der institutseigenen Datenbank mit einer individuellen Stammsammlungsnummer erfasst; tagesaktuell können die Anzahl an Lagergefäßen und die Lagerorte ausgelesen werden. Von nahezu allen Stämmen wurden 4 Gene komplett sequenziert (sie kodieren für das Hämagglutinin, das cytokine response modifier B Protein, das chemokine binding Protein und das Fusionsprotein); die Sequenzen sind in einer Datenbank hinterlegt. Mit seiner umfangreichen Stammsammlung ist das InstMikroBioBw auf dem Gebiet der Diagnostik der Pockenviren ein gefragter Partner. Dies hat zu Kooperationen mit dem CDC [7, 8] und Koltsovo (den beiden WHO-Referenzzentren für Pocken), dem RKI (Konsiliarlabor für Pocken) und anderen Partnern geführt [9 - 11].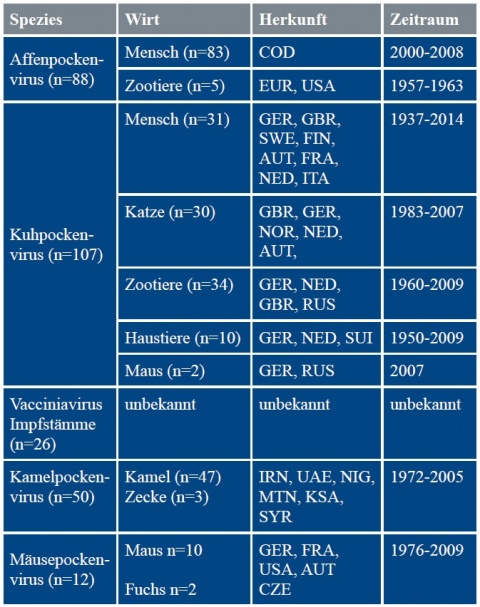
{kind=link}
Gen- und Genomsequenzierung
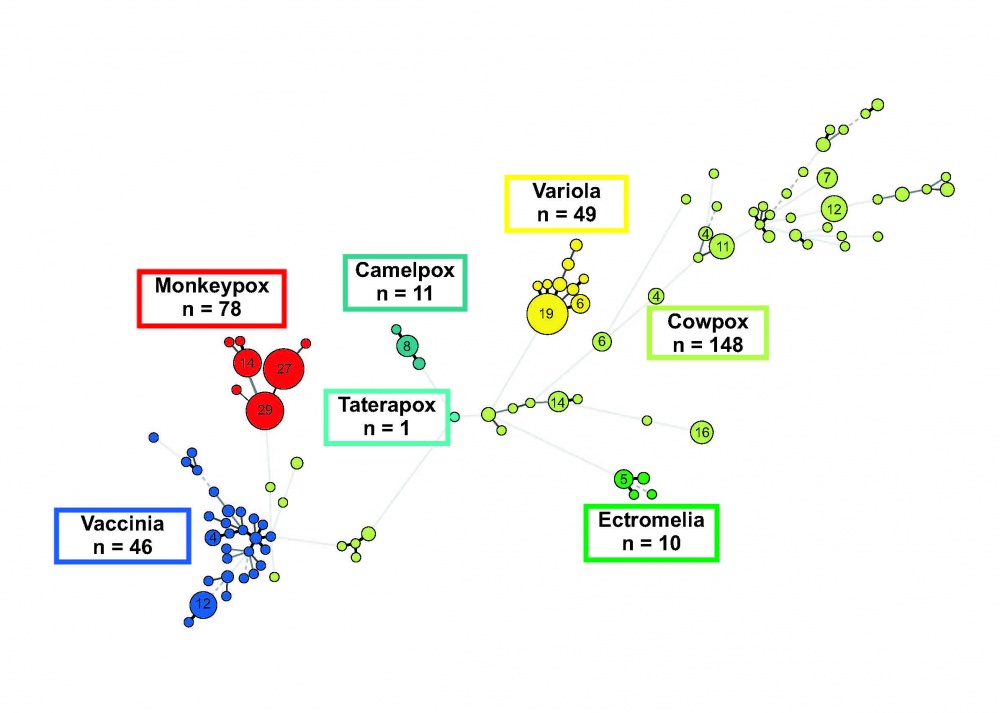
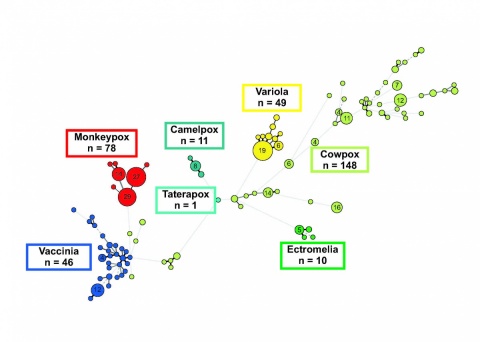
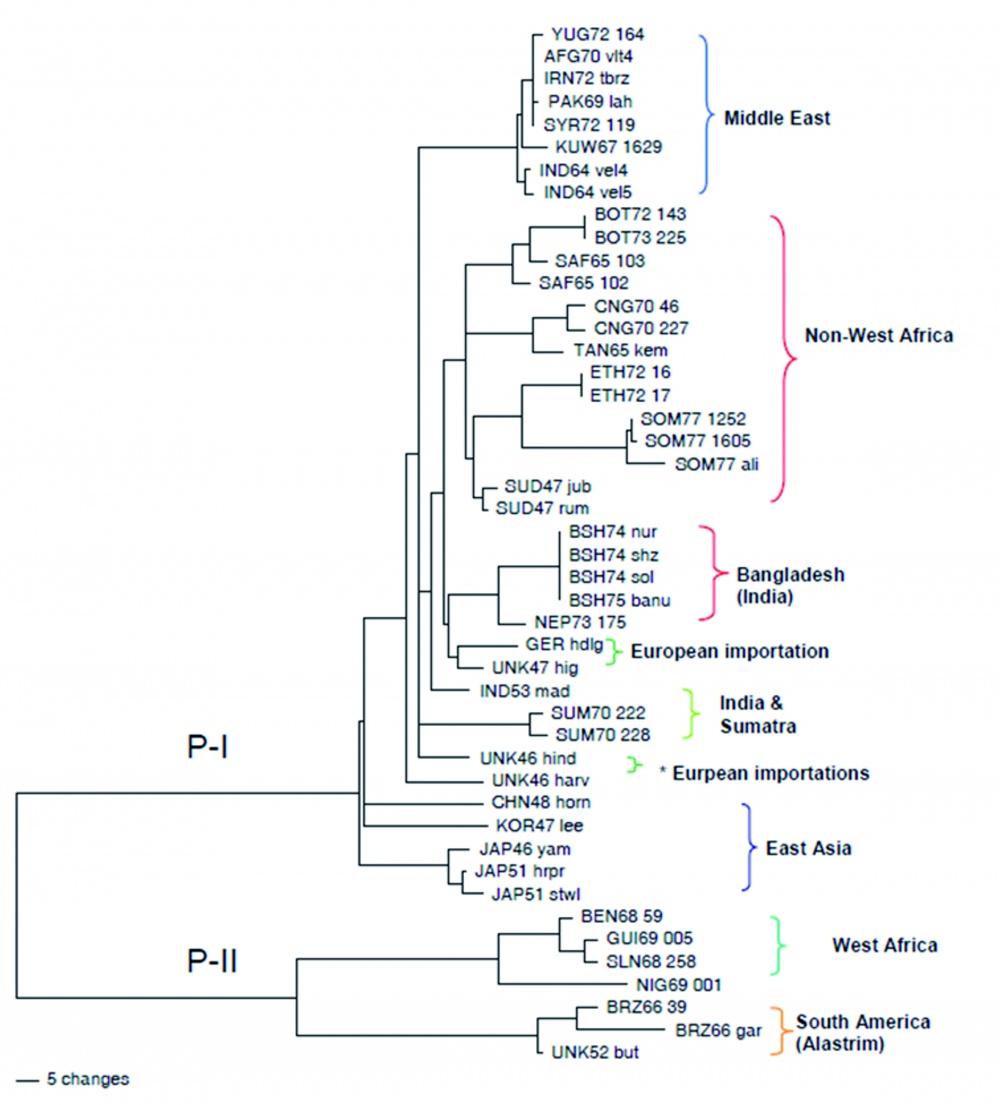
Die Sequenzierung von Genen - und in der letzten Zeit von Genomen - bestätigt die vor Jahrzehnten aufgrund phänotypischer Unterschiede im infizierten Brutei festgelegte taxonomische Unterteilung in die bekannten Orthopockenvirus-Spezies. Eine Ausnahme bilden die Kuhpockenviren. Deren genetische Diversität ist so groß, dass einige Forscher von mindestens zwei, andere dagegen von mehr als fünf Subtypen ausgehen. Die Bestimmung der Hämagglutinin-Sequenz mit etwa 1 000 Nukleotiden (das sind nur 0,5 % des Gesamtgenoms) reicht bei Kuhpockenviren aus, um nahezu jedes Isolat individuell zu unterscheiden. Dies ist in Abbildung 1 dargestellt. Sie zeigt einen phylogenetischen Datenbaum, der auf Hämagglutinin-Gensequenzen beruht. Gut zu erkennen ist, dass - mit Ausnahme der Kuhpockenviren - alle anderen Spezies nur eine geringe Sequenzvariation aufweisen. Variola major Stämme können zwar von Variola minor Stämmen unterschieden werden, ebenso Affenpockenviren aus Westafrika von denen aus Zentralafrika, eine weitere Differenzierung der einzelnen Variola- oder Affenpockenvirus-Stämme ist aber nicht möglich.
Ganz anders hingegen die Situation bei den Kuhpockenviren: Hier liegt eine außerordentlich hohe Diversität vor [12]. Diese Diversität ist hervorragend dazu geeignet, um Fragestellungen der molekularen Epidemiologie zu beantworten und Infektketten aufzuklären [13, 14]. Dies zeigt das Beispiel des Auftretens von Kuhpockenvirus-Infektionen beim Menschen -verursacht durch Schmuseratten. Beginnend ab Dezember 2008 traten in Deutschland, aber auch in Frankreich, Kuhpockenvirus-Infektionen beim Menschen auf, die offenbar durch infizierte „Schmuseratten“ verursacht worden waren. Von verschiedenen
{kind=link}
Stellenwert von Gesamtgenomsequenzierungen
Heutzutage begnügt man sich nicht mehr mit der Sequenzierung einzelner Gene; mittlerweile ist die Sequenzierung des Gesamtgenoms Stand der Technik. In Zusammenarbeit mit dem CDC und dem RKI werden derzeit etwa 60 Kuhpockenvirus--Stämme des InstMikroBioBw sequenziert. Diese Daten werden weiterführende Aussagen zur Mikroevolution sowie zum Genotyp ermöglichen. Schon im Jahre 2000 hatte man sich am CDC entschlossen, das Genom von 45 Variolavirus-Stämmen zu sequenzieren und - nach längerer politischer Diskussion - wurden diese Daten 2005 auch publik gemacht [17]. Damit waren Aussagen zur Phylogenie der Variolaviren möglich. Die 45 Stämme waren in der Zeit von 1942 bis 1976 isoliert worden und sind von der Herkunft und vom Krankheitsverlauf als repräsentativ anzusehen. Nach bioinformatischer Auswertung können zwei clades (P-I und P-II) unterschieden werden: clade P-II ist zweigeteilt und enthält die Alastrim oder Variola minor Stämme (Letalität unter 2 %) und Stämme aus Westafrika (Abbildung 2). Zur clade P-I gehören alle Stämme aus Asien mit Japan, China und Indien sowie aus Ost- und Südafrika, aber auch Stämme aus Europa, bei denen es sich aber nachweislich um Importe aus endemischen Ländern handelt.
Innerhalb der clade P-I können geographisch bzw. von einem Ausbruch her zusammengehörige Stämme differenziert werden. So sind Stämme aus
{kind=link}
weltweit getilgt sind, muss jedes Auftreten von Pocken als absichtliche Freisetzung angesehen und entsprechend verfolgt werden.
Kernaussagen/Fazit
• Die Freisetzung von Pockenviren hätte eine weltweite Gesundheitskatastrophe zur Folge.
• Bei Pockenverdacht ist die rasche und zuverlässige Identifizierung des Erregers der Schlüssel für alle Maßnahmen zur Eindämmung eines Ausbruchs.
• Untersuchende Labore tragen auf Grund der möglichen Folgen eine hohe nationale und internationale Verantwortung.
• Das InstMikroBioBw verfügt als akkreditiertes Institut über geeignete Untersuchungsverfahren zur raschen Diagnosestellung bei Pockenverdacht
Literatur
- Henderson DA, Inglesby TV, Bartlett JG et al.: Smallpox as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA (1999); 281 (22): 2127 - 2137.
- Drosten C: SARS. Weltreise eines neuen Virus. Biologie in unserer Zeit. Weinheim: Wiley-VCH 2003; 33(4): 212 - 213.
- Damon I, Meyer H and Shchelkunov S: Laboratory diagnostics for smallpox (Variola virus). In: Scientific review of variola virus research, 1999–2010. AS Khan and GL Smith (eds): WHO 2010; 17 - 36.
- Olson VA, Laue T, Laker MT et al.: Real-time PCR system for detection of orthopoxviruses and simultaneous identification of smallpox virus. J Clin Microbiol 2004; 42: 1940 - 1946.
- Nitsche A and Meyer H: Variola: Smallpox. in: BSL3 and BSL4 Agents. Epidemiology, Microbiology and Practical Guidelines. M. Elschner, S. Cutler, M. Weidmann, and P. Butaye, (eds): Wiley-Blackwell, Weinheim 2012; 201 - 210.
- Meyer H, Totmenin A, Shchelkunov S et al.: Variola and camelpox virus-specific sequences are part of a single large open reading frame identified in two German cowpox virus strains. Virus Research 2005; 108: 39 - 43.
- Carroll DS, Emerson GL, Li Y, et al.: Chasing Jenners Vaccine: Revisiting Cowpox virus Classification. PLoS ONE 2011; 6(8): e23086.
- Li Y, Meyer H, Zhao H et al.: GC content-based pan-pox universal PCR assays for poxvirus detection. J Clin Microbiol 2010; 48: 268 - 276.
- Maksyutov RA, Gavrilova EV, Meyer H et al.: Real-time PCR assay for specific detection of cowpox virus. J Virol Methods 2015; 211: 8 - 11.
- Duraffour S, Mertens B, Meyer H et al.: Emergence of cowpox: study of the virulence of clinical strains and evaluation of antivirals. PLoS ONE 2013; 8(2): e55808. Epub 2013 Feb 15.
- Rimoin AW, Mulembakanic PM, Johnston SC et al.: Major increase in human monkeypox incidence 30 years after smallpox vaccination campaigns cease in the Democratic Republic of Congo. PNAS USA 2010; 107(37): 16262 -16267. Epub 2010 Aug 30.
- Kurth A, Straube M, Kuczka A et al.: Cowpox Virus outbreak in Banded Mongooses (Mungos mungo) and Jaguarundis (Herpailurus yagouaroundi) with a time-delayed infection to humans. PLoS ONE 2009; 4(9): e6883.
- Kaysser P, von Bomhard W, Meyer H et al.: Genetic diversity of feline cowpox virus, Germany 2000–2008. Vet Microbiol 2010; 141: 282 - 288.
- Hemmer CJ, Littmann M, Loebermann M et al.: Human cowpoxvirus infection acquired from a circus elephant in Germany. Int J Infect Dis 2010; 14 Suppl 3: e338 – e340. Epub 2010 Jun 26.
- Campe H, Zimmermann P, Glos K et al.: Cowpox virus transmission from pet rats to humans, Germany. Emerg Infect Dis 2009; 15: 777 - 780.
- Ninove L, Domart Y, Vervel C et al.: Cowpox virus transmission from pet rats to humans, France. Emerg Infect Dis 2009; 15: 781 - 784.
- Esposito JJ, Sammons SA, Frace AM et al.: Genome sequence diversity and clues to the evolution of Variola virus. Science 2006; 313: 807 - 812.
Interessenkonflikt:
Die Autoren erklären, dass kein Interessenkonflikt besteht.
Für die Verfasser:
Oberstveterinär Prof. Dr. Hermann Meyer
Institut für Mikrobiologie der Bundeswehr
Neuherbergstr 11, 80937 München
E-Mail: [email protected]
.
[1]
Gemäß Biostoffverordnung werden Biostoffe in 4 Risikogruppen eingeteilt. Die Gruppe 3 umfasst solche Biostoffe, die eine schwere Krankheit beim Menschen hervorrufen und eine ernste Gefahr für Beschäftigte darstellen können; die Gefahr einer Verbreitung in der Bevölkerung kann bestehen, doch ist normalerweise eine wirksame Vorbeugung oder Behandlung möglich.“
Datum: 29.08.2016
Wehrmedizinische Monatsschrift 2016/6