NEUROENDOKRINE TUMOREN DES GASTROINTESTINALTRAKTES – DIAGNOSTIK, CHIRURGISCHE THERAPIESTRATEGIEN UND INTERDISZIPLINÄRES MANAGEMENT: FALLBERICHTE
Neuro-endocrine tumours of the gastrointestinal tract – diagnostics, surgical therapy strategies and interdisciplinary management: Case reports
Aus der Abteilung für Allgemein-, Viszeral- und Thoraxchirurgie¹ (Leitender Arzt: Oberstarzt Dr. F. Gatzka), der Abteilung Radiologie² (Leitender Arzt: Oberstarzt Dr. H. M. Schlegel), der Abteilung Laboratoriumsmedizin³ (Leitender Arzt: Flottillenarzt Dr. A. Fritsch) und der Abteilung Innere Medizin⁴ (Leitender Arzt: Oberstarzt Dr. C. Busch) am Bundeswehrkrankenhaus Hamburg (Chefarzt: Generalarzt Dr. J. Hoitz) sowie aus der Abteilung für Allgemein-, Viszeral- und Thoraxchirurgie⁵ (Chefarzt: Prof. Dr. K.-J. Oldhafer), Asklepiosklinik Barmbek
Lena Marie Heidelmann¹, Andreas Westerfeld¹, Leonid Kasakov¹, Christian Moritz², Franziska Puttnies³, Nicole Müller⁴, Karl-Jürgen Oldhafer⁵, Friedrich Gatzka¹ und Wilm Rost¹
WMM, 57. Jahrgang (Ausgabe 12/2013: S. 335-340)
Zusammenfassung
Hintergrund: Neuroendokrine Tumoren (NET) gehören zu den seltenen Primarien des Gastrointestinaltraktes. Beim Vorkommen im Darm werden sie oft erst in späten Stadien und beim Auftreten von Komplikationen diagnostiziert. Dies stellt hohe Anforderungen an Diagnostik und Therapie. Wir sahen in einem kurzen Zeitraum drei Patienten mit NET. Das veranlasste uns, die komplexe Problematik und interdisziplinäre Behandlungspfade zu erfassen.
Methoden: Literaturrecherche, Aufzeigen interner Handlungsabläufe und Therapiestrategien an drei Fallberichten.
Ergebnisse: NET des Darms, die erst durch eine Ileus-Symptomatik auffallen, differieren makroskopisch in der Regel nicht von anderen Tumorarten. Das chirurgische Vorgehen ist daher abhängig vom intraoperativen Befund.
Schlussfolgerungen: Therapiestrategien von NET sollten durch ein interdisziplinäres Team bestimmt und realisiert werden. Es sind weitere Studien erforderlich, um eine zuverlässige Diagnostik für ein früheres Aufspüren dieser Tumoren zu entwickeln
Schlagworte: Neuroendokrine Tumoren, NET, interdisziplinäres Management, Bundeswehrkrankenhaus Hamburg.
Summary
Background: Neuro-endocrine tumours (NET) are rare in having their primary source in the gastrointestinal tract. If located in the bowel they are often diagnosed in late stages and with appearance of one of its complications. High demands arise for diagnostics and therapy. We saw 3 patients with NET within a short period. This fact induced us to document the complexity of the disease and interdisciplinary therapy strategies.
Methods: Literature research, indicating of internal courses of action and therapy strategies based on 3 case reports.
Results: NET of the bowel which occur only by symptoms of an ileus do not differ from other kinds of tumours in macroscopic view. Therefore, the surgical procedures are depending on intra-operative results.
Conclusions: The therapy strategies of NET should be determined and implemented by an interdisciplinary team. More reliable diagnostic tests to track down these tumours more early should be developed in additional studies.
Keywords: Neuro-endocrine tumours, NET, interdisciplinary management, Bundeswehr hospital Hamburg.
Einleitung
Neuroendokrine Tumoren (NET) gehören generell mit einem Anteil von circa 0,49 % aller malignen Tumoren zu den seltener auftretenden Neoplasien, wobei Tumoren des Gastrointestinaltraktes gegenüber anderen Lokalisationen überwiegen (1–3).
Das deutsche NET-Register zeichnete die Lokalisationen von neuroendokrinen Primärtumoren in Deutschland auf, wobei 25,8 % dieser Neoplasien im Dünndarm sowie 3,7 % in der Appendix auftraten und 6,9 % sich im Bereich Colon/Rektum befanden (4). Vergleicht man diese Daten mit anderen Studien aus den Vereinigten Staaten von Amerika oder aus Asien und Europa, variieren sie leicht: Bezogen auf alle neuroendokrinen Tumoren, traten Tumoren des Rektums zu 7 – 27 %, des Dünndarms zu 20 – 25 %, des Colons zu 7 – 8 % und der Appendix zu 2 – 5 % auf (5–7). Zusätzlich würde die Inzidenz der in Tumorregistern gemeldeten Tumoren steigen (3, 5–7).
Der Ursprungsort von Zellen gastroenteropankreatischer neuroendokriner Tumoren (GEP-NET) wird unterschiedlich beschrieben. Eine Theorie besagt, dass es sich um verstreute Zellen des Gewebes der Neuralleiste, des Neuroektoderms, handeln würde. Andere Autoren beschreiben multipotente Stammzellen als Ursprung dieser Tumoren (8–10). Tumoren, die zum Beispiel im Rahmen eines Karzinoidsyndroms als funktionell anzusehen sind, werden von nicht-funktionellen Tumoren abgegrenzt und lösen auf Grund der unterschiedlichen produzierten Mediatoren eine heterogene Symptomatik aus (9, 11, 12).
Funktionell inaktive GEP-NET kommen häufiger vor, verhalten sich lange Zeit klinisch inapparent und werden oft erst durch Lokalkomplikationen wie Bauchschmerzen, Ileus, intestinale Ischämie, Ikterus oder gastrointestinale Blutungen erkennbar (11, 13). Daher werden sie häufig erst in späten Tumorstadien symptomatisch (11, 13).
Das klinische Erscheinungsbild funktionell aktiver Tumoren ist auf Grund der verschiedenen produzierten Metaboliten vielfältig (9, 11, 12). Zu den klinischen Erscheinungen solcher Neubildungen gehören beispielsweise: Stuhlunregelmäßigkeiten, abdominelle Schmerzsymptomatik, Ileus, Karzinoidsyndrom (mit Auftreten von zum Beispiel Flush-Symptomatik, abdominellen Beschwerden, Diarrhöen, Herzrhythmusstörungen, Fibrosierungen des Endokards) und B-Symptomatik (12–16).
Ziel dieser Arbeit war es, eine interdisziplinäre Handlungsanweisung zu erstellen und den ersten interdisziplinären, das heißt, kombinierten onkologischen, allgemeinchirurgischen, radiologischen, klinisch-chemischen und gastroenterologischen Behandlungspfad am Bundeswehrkrankenhaus Hamburg zu etablieren.
Methoden
Eine umfassende Literaturrecherche erfolgte über das Onlineportal von www.PubMed.org. Diese Ergebnisse wurden in einer interdisziplinären Konferenz vorgestellt und diskutiert. Hieraus ergaben sich interdisziplinäre Behandlungspfade mit Checklisten, welche in die intranet-gestützen Qualitäts-Handbücher der jeweiligen Abteilungen und in das allgemeine Qualitätsportal eingestellt wurden. Außerdem wurden eigene Behandlungserfahrungen einbezogen.
Ergebnisse
Diagnostik
Die notwendige Diagnostik und Therapierichtlinien neuroendokriner Neubildungen sind zuletzt 2012 in den Leitlinien der European Neuroendocrine Tumor Society (ENETS) für den europäischen Sprachraum festgelegt worden (17–23).
Zur Basisdiagnostik des Stagings gehören die Bestimmung von Chromogranin A (CgA) als Tumormarker und 5-Hydroxyindolessigsäure (5-HIES) im 24-h-Urin, des Serotonins im plättchenreichen Plasma und der neuronenspezifischen Enolase (NSE) als Tumormarker bei entdifferenzierten Tumoren (17, 21, 24, 25). Bei Tumoren des Colons/Rektums wird ferner eine Bestimmung des Beta-Anteils des humanen Choriongonadotropins (ß-HCG) empfohlen (17, 24, 25). Als Anhalt für eine Metastasierung – primärer Metastasierungsort ist die Leber – gelten klinisch ein Karzinoid-Syndrom und laborchemisch eine stark erhöhte 5-HIES im 24-h-Urin (normal bis 9 mg/d, > 15 mg/d hochsignifikant für GEP-NET, > 350 mg/d sind hoch metastasenverdächtig für GEP-NET) (9, 12, 17, 21, 22, 24, 25). Der First-pass-Effekt der Leber ist die Ursache für die häufig unauffällige Klinik bei gastrointestinalen NET ohne Metastasierung mit Primarius im Pfortadereinzugsgebiet (9, 12, 17, 21, 22, 24, 25).
Die Spezialdiagnostik bei funktionellen Tumoren umfasst die Echokardiografie und Bestimmung des diagnostischen Parameters für Herzinsuffizienz NT-proBNP (12, 21, 25).
Zum weiteren Staging werden generell eine abdominelle Sonografie (gegebenenfalls mit Kontrastmittel), eine Endosonografie, eine abdominelle Computertomografie (CT) beziehungsweise Magnetresonanztomografie (MRT) mit Kontrastmittel empfohlen (12, 17, 21, 24). Mögliche zusätzliche Untersuchungen können zur Primariusdiagnostik erforderlich sein, wie zum Beispiel eine Koloskopie oder Videokapsel-Endoskopie (12, 17, 21, 24). In Abhängigkeit von der Expression von Somatostatinrezeptoren beziehungsweise der proliferativen Aktivität des Tumors ist anstelle einer Somatostatin-Rezeptor-Szintigrafie (Octreotidscan) zur Metastasensuche eine 18F-Fluordeoxyglukose-Positronenemissionstomografie/Computertomografie (FDG-PET-CT) bei undifferenzierten NET und/oder negativem Octreotidscan eine 68-Gallinium-DOTATOC-Positronenemissionstomografie/Computertomografie (68-Ga-DOTATOC-PET-CT) mittels Edotreotid bei gut differenzierten NET mit Karzinoidsyndrom möglich (12, 17, 21, 24).
Histopathologisch sollte die oberflächliche Expression von Somatostatinrezeptoren (SSTR) der neuroendokrinen Zellen festgestellt werden, um die Bindungsaffinität von Somatostatinanaloga für eine spätere Therapieoption festlegen zu können (9, 27, 28).
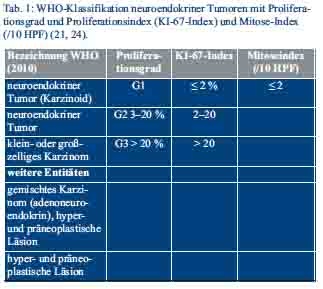
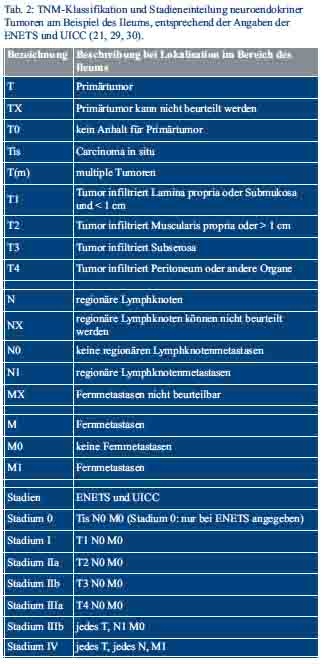
Die Einteilung der neuroendokrinen Tumoren erfolgt nach den Maßgaben der WHO-Klassifikation von 2010, inklusive eines proliferationsbasierten Gradings, der 7. TNM-Klassifikation und der Klassifikation der Union for International Cancer Control (UICC) (Tab. 1 und Tab. 2) (28–30).
Die Bestimmung der Proliferationsrate erfolgt am histopathologischen Präparat durch Festlegung des Ki-67-Index und des Mitose-Index, der anhand der ausgezählten Mitosen pro 10 Hauptgesichtsfelder (HPF) berechnet wird (24).
Therapie
Prognostische Faktoren und Therapiekonzepte richten sich nach der korrekten Tumorstadien-Einteilung, die vom Ausbreitungsgrad und von der Proliferationsaktivität des Tumors abhängen (24).
Chirurgische Resektionen von neuroendokrinen Tumoren sind entweder als optional kurative Eingriffe mit R-0-Resektionen oder als palliative Eingriffe im Sinne eines Tumor-Debulkings anzusehen (31). R1-Resektionen sollen die Prognose der Tumorerkrankung verbessern, wobei auch R-2-Resektionen manchmal das Ansprechen einer Chemotherapie erhöhen können (31).
Das chirurgische Management von NET des Ileums ist stark abhängig von einer Metastasierung und einem möglichen multifokalen Wachstum, das in 30 – 40 % der Fälle auftreten kann, und sollte so frühzeitig wie möglich erfolgen (9, 31, 32). Ein Ileusgeschehen führt laut Kaudel et al. in circa 30 % der Fälle zur Operation (31). Im Rahmen eines kurativen Ansatzes sollte zusätzlich zur Resektion des Primarius eine systematische mesenteriale Lymphadenektomie durchgeführt werden (31). Bei Patienten mit Tumoren des terminalen Ileums sollten nicht nur eine Dünndarmsegmentresektion, sondern auch eine rechtsseitige Hemikolektomie erfolgen, während bei proximalen Tumoren des Ileums eine Dünndarmsegmentresektion mit keilförmiger Entfernung des dazugehörenden Mesenteriums meistens als ausreichend angesehen wird (21, 31–33).
Tumoren der Appendix vermiformis < 1 cm sind im Rahmen einer Appendektomie meistens kurativ reseziert (21, 31). Patienten mit Tumoren der Appendixbasis oder Tumoren > 2 cm benötigen nicht nur eine Appendektomie, sondern auch eine Hemicolektomie rechts (21, 31). Bei Tumoren mit Grenzgrößen zwischen 1 – 2 cm Ausdehnung bleibt die Einzelfallentscheidung, da in der aktuellen Literatur keine klare Vorgabe definiert ist (16, 17).
Kolorektale neuroendokrine Tumoren sollten wie kolorektale Adenokarzinome versorgt werden, wobei eine Hemikolektomie beziehungsweise eine subtotale Kolektomie mit Lymphadenektomie angezeigt ist (17, 31). Zusätzlich wird empfohlen, das Vorgehen abhängig von Ausdehnung/Größe des Tumors zu machen, wobei als Landmarken Ausdehnungen von < 1 cm und > 2 cm bei gut differenzierten Tumoren beschrieben werden (16, 17).
Speziell am Rektum ist die endoskopische Entfernung von neuroendokrinen Tumoren, die nicht bis in das Gewebe der Muscularis propria reichen (T1-Tumoren), teilweise möglich. In den anderen Fällen sollte zum Beispiel eine anteriore Rektumresektion mit totaler mesorektaler Exzision angestrebt werden (17, 31).
Sind Lebermetastasen bei einem Patienten nachgewiesen, empfehlen einige Autoren die simultane Entfernung der Gallenblase. Hintergrund hierfür ist die mögliche Koinzidenz eines gehäuften Auftretens einer Cholezystitis bei begleitendem Gallensteinleiden (21, 22, 34). Auch eine therapeutische Gabe von Octreotid vor jeder interventionellen Untersuchung und/oder vor Operationen von neuroendokrinen Tumoren periinterventionell/perioperativ sollte zur Prävention einer möglichen Karzinoidkrise abgewogen werden (33).
Falldarstellungen
Fall 1
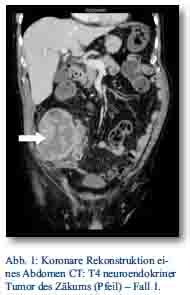
Im Oktober 2012 stellte sich ein 81-jähriger Patient mit progredienten rechtsseitigen Unterbauchschmerzen und Ileusbild in der zentralen Notaufnahme vor. Im durchgeführten CT wurde eine Darmpassagestörung sowie ein coecaler partiell nekrotischer und infiltrativer Tumor gesichert (Abb. 1). Aufgrund des klinischen Bildes wurde das Tumorgeschehen mittels Hemikolektomie rechts operativ versorgt. Das Staging mit Octreotidscan zeigte einen neuroendokrinen Tumor des Zäkums (pT4; pN0 (0/17); M0; G3; L0; V0) mit tumorfreien Resektionslinien (UICC IIIA), Ki-67-Index > 60 % (Abb. 3a/b). Es zeigte sich bei der Tumormarkerbestimmung ein CgA von 20 nMol/l (Normwert < 4), während Serotonin und NSE im Normbereich lagen. Die nach Entschluss des Tumorboards indizierte Chemotherapie wurde von dem Patienten vorerst abgelehnt. Er wurde im Verlauf beschwerdefrei entlassen. Bei wieder aufgetretener Ileussymptomatik und Verdacht auf ein Tumorrezidiv wurde der Patient jedoch im Dezember 2012 erneut aufgenommen und eine Dünndarmsegmentresektion durchgeführt. Hier konnte histologisch ein Lokalrezidiv nachgewiesen werden. Der Patient wurde daraufhin nach Portanlage durch die onkologische Abteilung unseres Hauses weiterbetreut und eine Chemotherapie mit Carboplatin/Etoposid eingeleitet.
Fall 2
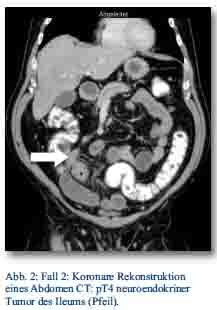
Im Oktober 2012 präsentierte sich ein 56-jähriger Patient mit gürtelförmigen Oberbauchschmerzen und Stuhlunregelmäßigkeiten in der zentralen Notaufnahme. Bei der weiteren Abklärung zeigte sich im CT des Abdomens (Abb. 2). und in der Koloskopie ein Tumor im Colon ascendens ohne Metastasennachweis, der in der Koloskopie nicht passierbar war.
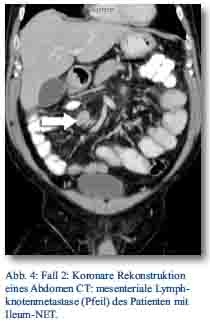
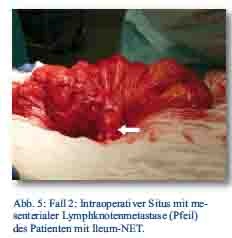
Daraufhin erfolgte bei gegebener Indikation eine onkologische Hemikolektomie rechts mit Ileotransversostomie. Das Staging ergab einen neuroendokrinen Tumor des Ileums (pT4; pN1 (5/12); M1; G1; L1; V1; Pn 1) mit tumorfreien Resektionslinien (UICC IV), Ki-67-Index circa 1 %. Die Tumormarker CgA (27 nMol/l; Normwert < 4 nMol/l) und Serotonin (480 mg/l; Normwert < 200 mg/l) waren stark erhöht und der NSE im Normbereich. Im Octreotidscan zeigte sich ein Verdacht auf eine mesenteriale Lymphknotenmetastase (Abb. 3a/b). Im Januar 2013 erfolgte dann die erneute stationäre Aufnahme zur offenen Entfernung der möglichen Lymphknotenmetastase, die zusätzlich auch CT-diagnostisch abgeklärt wurde (Abb. 4). Bei der intraoperativen Organdurchsicht zeigte sich neben der Lymphknotenmetastase (Abb. 5) eine suspekte Veränderung im Segment VI der Leber. Daraufhin konnte diese atypisch exzidiert werden (Abb. 6). Postoperativ wurden der Lymphknoten histologisch als metastatisch entartet und die Veränderung des Segments IV der Leber als Metastase identifiziert. Im MRT ließen sich daraufhin zusätzlich im Segment V Metastasen nachweisen. Es erfolgte dann im Februar 2013 eine atypische Leberresektion unter Einschluss der Metastasen des Segments V mit Zuhilfenahme von intraoperativer Sonografie in Zusammenarbeit mit Prof. Dr. Oldhafer, Chefarzt der Asklepiosklinik Barmbek (Abb. 7). Danach wurde der Patient in eine ambulante Nachsorge übergeben. Aktuell wird dort eine chemotherapeutische Behandlung nicht erwogen.
Fall 3
Im Dezember 2012 stellte sich ein 27-jähriger Patient mit progredienten rechtsseitigen Schmerzen im Mittel- und Unterbauch und klinischen Zeichen einer Appendizitis in der zentralen Notaufnahme vor. Sonografisch lag ein Konglomerat im rechten Unterbauch vor. Daraufhin wurde bei Verdacht auf Appendizitis eine laparoskopische Appendektomie durchgeführt. Intraoperativ zeigte sich die Appendix an der Spitze perforiert und bei Palpation verhärtet. In der histopathologischen Untersuchung wurde ein neuroendokriner Tumor der Appendixspitze diagnostiziert, pT1b; N0; M0; G1; L0; V0 mit tumorfreien Resektionslinien (UICC I), Ki-67-Index ca. 1 % (Abb. 8a/b). Bei Befundeingang wurde der Patient über das Tumorleiden informiert und zum Staging im Januar 2013 erneut stationär aufgenommen. Der Tumormarker CgA war mit 6 nMol/l erhöht, während NSE im Normbereich lag. Das weitere Staging inklusive Octreotidscan war unauffällig. Aufgrund der Leitlinien wurde kein weiteres chirurgisches Vorgehen angeraten und der Patient wurde eine ambulante onkologische Weiterbetreuung entlassen.
Interdisziplinäres Management
Bei einem Nachweis eines neuroendokrinen Tumors ist es, unabhängig von der Diagnose stellenden Abteilung, unabdingbar, eine interdisziplinäre Tumorkonferenz durchzuführen, um für den Patienten Therapiebausteine und weitere diagnostische Schritte festzulegen. Davor besteht die Möglichkeit, im Intranet anhand des ersten interdisziplinären Behandlungspfades des Bundeswehrkrankenhauses (BwKrhs) Hamburg, der jeweils im Qualitätshandbuch der Abteilungen Chirurgie und Innere hinterlegt ist, diagnostische Schritte einzuleiten. Die konsiliarische Mitbeurteilung der Patienten durch mehrere Fachdisziplinen unseres Hauses wie die Onkologie, Gastroenterologie, Kardiologie, Radiologie und Chirurgie gehören zum Standardprozedere. Von chirurgischer Seite ist ein Vorgehen nach den aktuellen Leitlinien der ENETS anzustreben. In unserer Abteilung für Allgemein-, Viszeral- und Thoraxchirurgie werden eine frühzeitige kurative Resektion unabhängig von der Tumorgröße und falls nötig eine kurative Nachresektion mit Lymphadenektomie, zum Beispiel bei vorerst endoskopisch entfernten Tumoren, favorisiert. Zusätzlich wird eine onkologische Resektion mit Tumorreduktion empfohlen. Eine Ileus-Symptomatik stellt grundsätzlich eine OP-Indikation dar. Sollte sich ein chirurgischer Handlungsbedarf ergeben, wird der Patient auf die allgemeinchirurgische Station aufgenommen und das weitere operative Prozedere geplant. Nach dessen Abschluss wird der Patient durch die onkologische Abteilung unseres Hauses weiter betreut und behandelt.
Schlussfolgerungen
Die Entwicklung eines interdisziplinären Behandlungspfades erwies sich als sinnvoll, um bei diesem komplexen, aber seltenen neuroendokrinen gastrointestinalen Tumoren den beteiligten Abteilungen die aktuelle Literatur verständlich und kurz gefasst und mit Empfehlungen darstellen zu können. Die Festlegung eines Behandlungspfades erscheint insbesondere dann sinnvoll, wenn es um eine komplexe Therapie von seltenen Tumorentitäten geht. In den hier vorliegenden Fällen wurden alle Patienten über die zentrale Notaufnahme unseres Hauses einer chirurgischen Betreuung zugeführt und mussten im Anschluss komplex onkologisch behandelt werden. Eine Standardisierung der Behandlungsstrategien nach der endgültigen Diagnosestellung ist aus unserer Sicht unumgänglich für das Outcome der Patienten. Aufgrund der zufälligen Kumulation des an sich seltenen Tumors in den letzten 12 Monaten wurden nun Behandlungspfade entwickelt, um die Abteilungsexpertisen dauerhaft zu erhalten und Staging-Strategien zu strukturieren.
Bildnachweis:
Folgende Fotos überließen freundlicherweise:
Abb. 1, 2, 4: Oberfeldarzt Dr. Moritz, Abteilung für Radiologie am Bundeswehrkrankenhaus Hamburg.
Abb. 3 a/b: Prof. Dr. Bohuslavizki, Nuklearmedizin Spitaler Hof, Spitalerstraße 8, 20095 Hamburg.
Abb. 5, 6: Flottillenarzt Dr. Rost, Abteilung für Allgemein-, Viszeral- und Thoraxchirurgie, Bundeswehrkrankenhaus Hamburg.
Abb. 7: Prof. Dr. K. J. Oldhafer, Chefarzt der Abteilung für Allgemein-, Viszeral- und Thoraxchirurgie, Asklepiosklinik Barmbek, Rübenkamp 220, 22291 Hamburg.
Abb. 8 a/b: Oberstarzt Dr. Göller, Leiter der Abteilung für Pathologie BundeswehrZentralkrankenhaus Koblenz, Rübenacher Straße 170, 56072 Koblenz.
Literatur
- Godwin JD, 2nd: Carcinoid tumors. An analysis of 2,837 cases. Cancer 1975; 36: 560–569.
- Modlin IM, Lye KD, Kidd M: A 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003; 97: 934–959.
- Modlin IM, Oberg K, Chung DC, et al.: Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 2008; 9: 61–72.
- Begum N, Maasberg S, Plockinger U, et al.: Neuroendocrine Tumours of the GI Tract - Data from the German NET Registry. Zentralbl Chir 2012; 137: e1.
- Hauso O, Gustafsson BI, Kidd M, et al.: Neuroendocrine tumor epidemiology: contrasting Norway and North America. Cancer 2008; 113: 2655–2664.
- Tsai HJ, Wu CC, Tsai CR, et al.: The epidemiology of neuroendocrine tumors in taiwan: a nation-wide cancer registry-based study. PLoS One 2013; 8: e62487.
- Yao JC, Hassan M, Phan A, et al.: One hundred years after „carcinoid“: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008; 26: 3063–3072.
- Barker N, van Es JH, Kuipers J, et al.: Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007; 449: 1003–1007.
- Kloppel G, Anlauf M: Epidemiology, tumour biology and histopathological classification of neuroendocrine tumours of the gastrointestinal tract. Best Pract Res Clin Gastroenterol 2005; 19: 507–517.
- Oberstein PE, Saif MW: Update on prognostic and predictive biomarkers for pancreatic neuroendocrine tumors. JOP 2012; 13: 368–371.
- Schmid RM: Neuroendokrine Tumoren. Der Onkologe 2011; 17: 570–571.
- Noe S, Mayr M, Scheidhauer K, et al.: Klinik und Diagnostik neuroendokriner Neoplasien. Der Onkologe 2011; 17: 583–591.
- Scherubl H, Jensen RT, Cadiot G, et al.: Neuroendocrine tumors of the small bowels are on the rise: Early aspects and management. World J Gastrointest Endosc 2010; 2: 325–334.
- Bhattacharyya S, Toumpanakis C, Chilkunda D, et al.: Risk factors for the development and progression of carcinoid heart disease. Am J Cardiol 2011; 107: 1221–1226.
- de Herder WW: Tumours of the midgut (jejunum, ileum and ascending colon, including carcinoid syndrome). Best Pract Res Clin Gastroenterol 2005; 19: 705–715.
- Hotz HG, Bojarski C, Buhr HJ: [Neuroendocrine colorectal tumors. Surgical and endoscopic treatment]. Chirurg 2011; 82: 607–611.
- Caplin M, Sundin A, Nillson O, et al.: ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: colorectal neuroendocrine neoplasms. Neuroendocrinology 2012; 95: 88–97.
- Delle Fave G, Kwekkeboom DJ, Van Cutsem E, et al.: ENETS Consensus Guidelines for the management of patients with gastroduodenal neoplasms. Neuroendocrinology 2012; 95: 74–87.
- Falconi M, Bartsch DK, Eriksson B, et al.: ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology 2012; 95: 120–134.
- Jensen RT, Cadiot G, Brandi ML, et al.: ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology 2012; 95: 98–119.
- Pape UF, Perren A, Niederle B, et al.: ENETS Consensus Guidelines for the management of patients with neuroendocrine neoplasms from the jejuno-ileum and the appendix including goblet cell carcinomas. Neuroendocrinology 2012; 95: 135–156.
- Pavel M, Baudin E, Couvelard A, et al.: ENETS Consensus Guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology 2012; 95: 157–176.
- Salazar R, Wiedenmann B, Rindi G, et al.: ENETS 2011 Consensus Guidelines for the Management of Patients with Digestive Neuroendocrine Tumors: an update. Neuroendocrinology 2012; 95: 71–73.
- Anlauf M, Gerlach P, Raffel A, et al.: Neuroendokrine Neoplasien des gastroenteropankreatischen Systems. Der Onkologe 2011; 17: 572–582.
- Thomas L: Labor und Diagnose : Indikation und Bewertung von Laborbefunden für die medizinische Diagnostik Bd. 1–14.4. Labordiagnostik gastrointestialer neuroendokriner Tumoren. 8. Aufl. ed. Frankfurt/Main: Th-Books Verl.-Ges.; 2012.
- Thomas L: Labor und Diagnose : Indikation und Bewertung von Laborbefunden für die medizinische Diagnostik Bd. 1–14.4. Labordiagnostik gastrointestialer neuroendokriner Tumoren. 8. Aufl. ed. Frankfurt/Main: Th-Books Verl.-Ges.; 2012.
- Anlauf M: Neuroendocrine neoplasms of the gastroenteropancreatic system: pathology and classification. Horm Metab Res 2011; 43: 825–831.
- Kloppel G: Classification and pathology of gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer 2011; 18 Suppl 1: S1–16.
- Kloppel G, Rindi G, Perren A, et al.: The ENETS and UICC TNM classification of neuroendocrine tumors of the gastrointestinal tract and the pancreas: comment. Pathologe 2010; 31: 353–354.
- Wittekind C, International Union against Cancer: TNM : Klassifikation maligner Tumoren. 7. Aufl., 4. korr. Nachdr. ed. Weinheim: Wiley-Blackwell; 2012.
- Kaudel CP, Watzka FM, Musholt TJ: Chirurgische Therapie gastroenteropankreatischer neuroendokriner Neoplasien. Der Onkologe 2011; 17: 609–620.
- Fendrich V, Bartsch DK: Surgical treatment of gastrointestinal neuroendocrine tumors. Langenbecks Arch Surg 2011; 396: 299–311.
33. Akerstrom G, Hellman P, Hessman O, et al.: Management of midgut carcinoids. J Surg Oncol 2005; 89: 161–169.
34. Eriksson J, Stalberg P, Nilsson A, et al.: Surgery and radiofrequency ablation for treatment of liver metastases from midgut and foregut carcinoids and endocrine pancreatic tumors. World J Surg 2008; 32: 930–938.
Anschrift für die Verfasser:
Dr. med. Lena Marie Heidelmann, Stabsarzt
Bundeswehrkrankenhaus Hamburg
Abteilung II: Allgemein-, Viszeral- und Thoraxchirurgie
Lesserstraße 180, 22049 Hamburg
E-Mail: lenamarieheidelmann@bundeswehr.org
Der Beitrag wird im Internet unter www.wehrmed.de publiziert.




Datum: 17.12.2013
Quelle: Wehrmedizinische Monatsschrift 2013/12