ALLES NUR AUFGREGUNG? RELEVANZ DER ABKLÄRUNG EINER BELASTUNGSHYPERTONIE: FALLBERICHT
Only excitement? On the relevance of addressing exercise-induced arterial hypertension: Case report
Aus dem Sanitätszentrum Nienburg, Truppenarztsprechstunde¹ (kommissarischer Leiter: Oberstabsarzt C. Lambrecht) und der Fachuntersuchungsstelle I des Fachsanitätszentrums Hannover² (Leiter: Oberstarzt Dr. K. Kilian)
Marc Potinius¹, Sebastian Scholz² und Klaus Kilian²
WMM, 58. Jahrgang (Ausgabe 1/2014: S. 22-25)
Zusammenfassung
Angeborene (genetische) Krankheiten gehören zumeist zu den seltenen Erkrankungen, die jedoch einen unterschätzten Einfluss auf die Lebensqualität, Letalität und Morbidität der Patienten haben. Daher sollten Allgemeinmediziner beim Ausschluss sekundärer Ursachen für häufige Erkrankungen wie Bluthochdruck auch an seltene Krankheiten denken. Angeborene Nierenkrankheiten wie die autosomal-dominante polyzystische Nierenerkrankung können beispielsweise zuerst mit einem erhöhten Blutdruck symptomatisch werden und sollten durch eine Abdomensonographie und Laboruntersuchungen ausgeschlossen werden.
Schlagworte: Arterielle Hypertonie, Ergometrie, genetische Erkrankung, Nierenzysten, ADPKD.
Summary
Hereditary diseases are orphan diseases with an underestimated health burden concerning their impact on patient´s live quality, mortality and morbidity. Therefore general practitioners should take into account orphan diseases while excluding secondary reasons for major health problems such as arterial hypertension. For example hereditary renal diseases as the autosomal dominant polycystic kidney disease (ADPKD) might first become symptomatic with elevated blood pressure and are recommended to be screened for with abdominal ultrasound as well as laboratory investigation.
Keywords: arterial hypertension, ergometry, hereditary disease, renal caysts, ADPDK
Einführung
Zu einer der truppenärztlichen Routineuntersuchungen im Rahmen von Begutachtungen auf Dienst-, Eignungs- und Verwendungsfähigkeit gemäß „Allgemeinem Umdruck 80“ gehört in Abhängigkeit der geforderten Verwendungsreihe eine Ergometrie. Diese wird nach entsprechender Patientenaufklärung durch dafür geschultes und auf das jeweilige Gerät eingewiesenes Assistenzpersonal durchgeführt.
Ein häufiger Abbruchgrund bei jungen Patienten ist, neben der muskulären Erschöpfung durch Überschreiten der aerob-anaeroben Schwelle und sich daraus entwickelnder Laktatazidose, das Erreichen des Blutdruck-Abbruchkriteriums. Hierzu werden je nach Literatur verschiedene Grenzwerte von > 230 – 280/115 – 130 mm Hg angegeben (1). Welche weiteren diagnostischen Schritte sollten bei Feststellung einer arteriellen Belastungshypertonie durch den Truppenarzt eingeleitet bzw. durchgeführt werden?
Fallbeschreibung
Am 1. August 2012 wurde im Sanitätszentrum ein damals 25-jähriger, bis dato gesunder, Obermaat mittels Ergometrie im Rahmen einer Begutachtung auf Ausland- und Tropendienstverwendungsfähigkeit untersucht. Die geforderte Mindest-PWC (physical working capacity) hierfür betrug 2,3 W/kg KG. Der Patient war 170 cm groß und wog 73,0 kg. Der Body Mass Index (BMI) lag bei 25,6 kg/m2. Er hatte bereits in Ruhe einen leicht erhöhten Blutdruck (RR) von 150/90 mm Hg. Im 12-Kanal-EKG zeigte sich eine Sinustachykardie mit einer Herzfrequenz von 118/min bei ansonsten unauffälligem Stromkurvenverlauf. Zeichen einer Rechtsherzbelastung und Hypertrophiezeichen fanden sich nicht. Der Auskultationsbefund war ebenfalls unauffällig. Die Ergometrie ergab unter Belastung einen raschen Anstieg des Blutdruckes, welcher zu einem Untersuchungsabbruch bei einer Last von 200 Watt führte. Dies entsprach einer PWC von 1,8 W/kg KG. Bei einer Last von 100 Watt wurde bereits ein RR > 200/100 mm Hg gemessen, womit per definitionem eine Belastungshypertonie vorlag. Nach Belastungsende normalisierten sich die Werte rasch bis auf 130/80 mm Hg.
Der Patient wurde daraufhin in die truppenärztliche Sprechstunde zur weiterführenden Anamneseerhebung und körperlichen Untersuchung einbestellt. Die Eigenanamnese war hinsichtlich kardiovaskulärer Risikofaktoren unauffällig. Er war Nichtraucher, verneinte regelmäßigen Alkoholkonsum glaubhaft und nahm keinerlei Medikamente, Nahrungsergänzungsmittel oder Muskelaufbaupräparate ein. Eine Ende Juli durchgeführte Blutuntersuchung hatte ein bis auf eine Eosinophilie – bei bekannter Rhinitis allergica – unauffälliges Blutbild, niedrig normale Transaminasen und normwertige Retentionsparameter sowie Elektrolyte ergeben. Die Stoffwechsellage war euthyreot. Auch ein Urin-Stix mittels semiquantitativem Combur-Test war unauffällig ausgefallen. Der Patient hatte keine bekannten Herz-, Lungen-, Nieren- oder Schilddrüsenerkrankungen und fühlte sich im Alltag und beim Dienstsport körperlich gut belastbar.
Die Familienanamnese ergab, dass der heute 59-jährige Vater des Patienten an einer Nierenerkrankung litt und bereits präventiv einen Shunt zur Dialysevorbereitung erhalten hatte. An die genaue Diagnose konnte der Patient sich nicht erinnern, „allerdings sei der Vater mit 46 Jahren wegen Zysten an den Nieren operiert worden.“ Die aus Thailand stammende Mutter war an „Altersdiabetes“ erkrankt. Der Patient hatte einen zwei Jahre jüngeren, gesunden Bruder. Bei diesem war bis dato keine Ultraschalluntersuchung der Nieren durchgeführt worden.
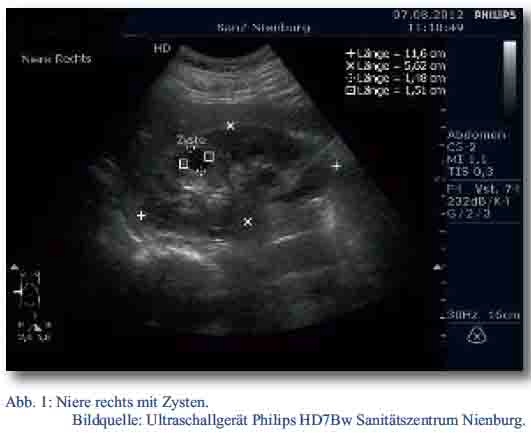
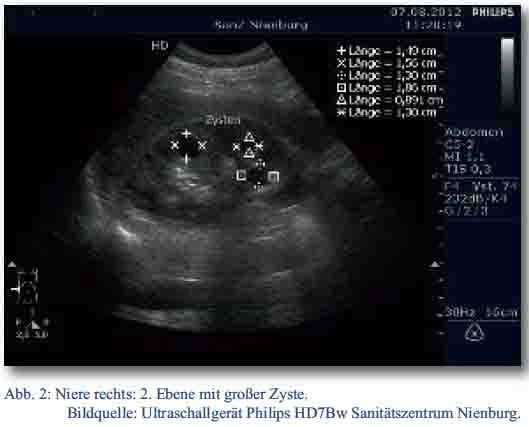
Der Patient wurde daraufhin sonographisch mittels des zu diesem Zeitpunkt neu in den truppenärztlichen Bereich eingeführten Ultraschallgerätes Philipps HD7Bw untersucht. Dabei fanden sich in der rechten Niere drei echofreie runde, glatt begrenzte Raumforderungen (RF) im Mark-Rindenbereich. Die größte RF maß circa 15 x 15 x 15 mm (Abb. 1 und Abb. 2). In der linken Niere konnte zunächst keine zystische Formation nachgewiesen werden. Ansonsten waren die Nieren sonographisch unauffällig.
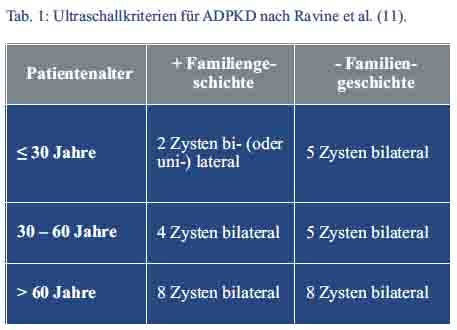
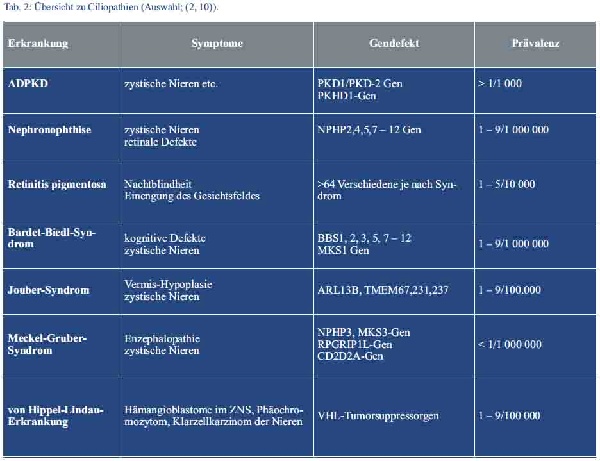
Aus der Anamnese und den sonographisch erhobenen Befunden ergab sich der Verdacht auf das Vorliegen einer autosomal-dominant erblichen polyzystischen Nierenerkrankung (siehe Tab. 1).
Der Patient wurde zur Bestätigung und weiterführenden Untersuchung an die Fachuntersuchungsstelle I des Fachsanitätszentrums Hannover überwiesen. Dort erfolgte eine erneute Sonographie des Abdomens. Dabei fand sich eine weitere, die Zystenkriterien erfüllende RF in der linken Niere. Zystische Formationen in Pankreas und Leber konnten nicht nachgewiesen werden.
Krankheitsbild der ADPKD
Die autosomal-dominant erbliche Variante der polyzystischen Nierenerkrankung (ADPKD) gehört zur Gruppe der Ciliopathien (Übersicht in Tab. 2). Sie stellt daher eine Multisystemerkrankung dar, bei welcher der in allen Zellen vorkommende Cilien-Centrosomen-Komplex betroffen ist (2). Ursächlich liegt bei 85 % der Betroffenen eine Mutation im PKD1- und bei 15 % eine Veränderung im PKD2-Gen vor, die entscheidend an der Differenzierung der renalen Tubuluszellen beteiligt sind (3). Das normale Genprodukt sind zwei Membranproteine, die zusammen einen in Cilien vorkommenden Ionenkanal aus der Subfamilie der TRP-Ionenkanäle bilden. Mit einer Prävalenz von 1 : 800 ist die ADPKD eine der häufigsten erblichen Erkrankungen mit autosomal-dominantem Erbgang (3). Für das Entstehen von Zysten ist auf zellulärer Ebene vermutlich eine somatische Spontanmutation im zweiten Allel (Second-hit-Hypothese) vonnöten. Die Zysten entstehen, weil die defekten Zilien, die normalerweise durch ihre mechanosensiblen Eigenschaften die planare Zellpolarität regulieren, abknicken und es so zu einer fehlerhaften Informationsweitergabe über den wahren Tubuluszellverlauf kommt. Das orientierte Zellwachstum wird somit verhindert und statt eines funktionstüchtigen Tubulusschlauches bilden sich Zysten.
Typischerweise wird die Erkrankung erst im jüngeren Erwachsenenalter aufgrund des progressiven Zystenwachstums und damit einhergehender Komplikationen wie Flankenschmerzen, Zystenruptur und -einblutung oder Makrohämaturie mittels Ultraschall diagnostiziert. Die Erkrankung führt meistens zu bilateralen Nierenzysten, wobei auch Zysten in anderen Organen wie Leber (fast in 100 % im Verlauf) und Pankreas (etwa in 10 %) assoziiert sind. Anlageträger haben ferner ein erhöhtes Risiko für intrakranielle Aneurysmata und bei 25 % der Betroffenen finden sich kardiologische Veränderungen wie eine Aortenektasie oder ein Mitralklappenprolaps. Es finden sich gehäuft intestinale Divertikel.
Eine 2009 durch Pei et al (4) publizierte Untersuchung löste die noch häufig angewandten Ravine-Kriterien (9) für die sonographische Diagose der ADPKD ab (Tab. 1). Seither genügt bereits der Nachweis von drei uni-/oder bilateralen oder zwei Zysten pro Niere bei Patienten < 59 Jahre und mit familiärer Vorbelastung (auch bei unbekanntem Genotyp) für die Diagnosestellung. Bei Patienten < 30 Jahre lag die Spezifität für die Diagnose einer ADPKD bei ≥ 3 uni-/bilateralen Zysten bei 100 %. Die Zystengröße hängt vom betroffenen Gen ab. Liegt eine PKD-1-Mutation vor, sind die Zysten größer und zu einem früheren Zeitpunkt nachweisbar als bei PKD-2-Mutationen (6).
Die Hälfte der von ADPKD betroffenen Patienten wird im Alter von 50 – 60 Jahren dialysepflichtig, wobei Patienten mit Mutationen im PKD1-Gen durchschnittlich 20 Jahre eher auf eine Nierenersatztherapie angewiesen sind, als jene mit Mutationen im PKD-2-Gen (5). Vorher kommt es zu einer kompensatorischen Hypertrophie der noch funktionstüchtigen Nephrone. Der Anteil von ADPKD-Patienten an der Gesamtheit aller dialysepflichtigen Patienten in der westlichen Welt beträgt in etwa 7 – 10 %. Damit stellt die ADPKD die im Krankheitsverlauf vierthäufigste zur Dialyse führende Nierenerkrankung dar. Eine Peritonealdialyse (PD) gilt zwar noch als relative Kontraindikation, in retrospektiven Studien und Registerdaten zeigte sich allerdings bis auf eine erhöhte Rate an Hernien kein Unterschied bezüglich Gesamtüberleben, Peritonitisrate und Effektivität (7). In den neuen Versorgungsleitlinien für Nierenerkrankungen bei Diabetes wird die PD mittlerweile als Initialbehandlung gegenüber der Hämodialyse (HD) favorisiert.
Die therapeutischen Möglichkeiten bei ADPKD sind begrenzt. In klinischen Studien durchgeführte Versuche einer Progressionsverzögerung des Zystenwachstums mittels Gabe von mTOR-Inhibitioren wie Sirolimus verliefen frustran (Suisse-ADPDK-Studie), obgleich die aberrante Aktivierung der mTOR-Signalkaskade bei der ADPKD in vorangegangenen Untersuchungen mit einer progressiven Nierengrößenzunahme assoziiert war (5).
Auch die Gruppe der Vasopressin V2-Rezeptor-Antagonisten konnte mit dem Vertreter Tolvaptan in der TEMPO-3:4-Studie nicht überzeugen. Zwar wurde eine Zunahme des Nierenvolumens und eine Abnahme der GFR über einen Zeitraum von drei Jahren verzögert, aufgrund des Nebenwirkungsspektrums und einer damit einhergehenden Non-Adhärenz von 23 % kommt eine Dauertherapie aber vermutlich nicht infrage (6). Zudem ist kein Effekt auf Leberzysten zu erwarten, da diese keinen V2-Rezeptor exprimieren.
Klinischer Verlauf
Dem Patienten wurde nach genetischer Beratung ein Gentest angeboten. Dabei fand sich die heterozygote Nonsense-Mutation c.2614C>T (p.Arg872X) im Exon 14 des PKD2-Gens. Diese Mutation ist bereits in der Literatur als pathogen beschrieben und wird in der PKD-Datenbank (http://pkd.mayo.edu) geführt. Sie verursacht einen frühzeitigen Translationsstopp und führt daher mit hoher Wahrscheinlichkeit zu einem Funktionsverlust des entsprechenden Proteins.
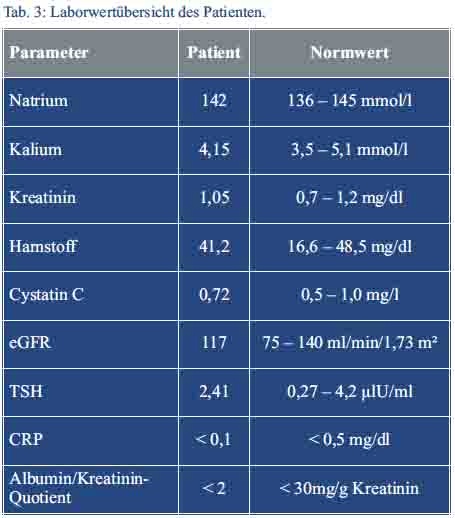
Zerebrale Aneurysmata konnten erfreulicherweise in einer cMRT ausgeschlossen werden. Die empfohlene 24-h-Sammelurinmessung zur Bestimmung der Kreatinin-Clearance wurde aufgrund der möglichen Probenfehler im ambulanten Setting durch die Bestimmung des Cystatin-C im Serum ersetzt. Hieraus konnte eine normale glomeruläre Filtrationsrate von 117 ml/min/1,73 m² ermittelt werden. Die Albumin/Kreatinin-Ratio zum Ausschluss einer Proteinurie war blande (Tab. 3). Eine 24-h-Langzeitblutdruckmessung zeigte durchweg normotensive Werte mit erhaltener zirkadianer Rhythmik.
Dem Patienten wurde empfohlen sich allgemein gesund zu verhalten, auf eine gesunde und ausgewogene Ernährung, ausreichende Trinkmengen und regelmäßige sportliche Aktivität zu achten. Ferner wurde Nikotinverzicht und allenfalls moderater Salz- und Alkoholkonsum empfohlen. Der Impfstatus wurde komplettiert und jährliche Grippeschutzimpfungen empfohlen. Über die nephrotoxische Wirkung, insbesondere von NSAR, ASS und Aminoglykosid-Antibiotika wurde aufgeklärt und Ausweichpräparate genannt. In Absprache mit dem Patienten wurde auf die primärprophylaktische Einnahme eines ACE-Hemmers verzichtet. Es wurden anfangs halbjährliche Wiedervorstellungen vereinbart.
Der Patient wurde zudem an die Sprechstunde einer nephrologischen Universitätsambulanz angebunden.
Schlussfolgerungen
Die sorgfältige Anamnese und körperliche Untersuchung im Rahmen der militärärztlichen Begutachtung eröffnet ein präventivmedizinisches Fenster. Chronische Erkrankungen können so frühzeitig erkannt und noch vor der Manifestation von Organschäden weiter abgeklärt und behandelt werden.
Auch vermeintlich harmlose Befunde wie eine Belastungshypertonie können Ausdruck der Erstmanifestation einer Organerkrankung sein und sollten abgeklärt werden.
Das in die truppenärztliche Ausstattung eingeführte Ultraschallgerät Philipps HD7Bw bietet bei entsprechender Anwenderausbildung eine hervorragende Möglichkeit zur frühen Detektion pathologischer Organveränderungen. Die flächendeckende Einführung sichert eine bestmögliche Befundkorrelation zwischen Truppenarzt und Fachuntersuchungsstelle.
Interessenkonflikt: Die Verfasser geben an, dass kein Interessenkonflikt besteht.
Literatur
- Wilson PD: Polycystic Kidney Disease. N Engl J Med 2004; Band 350: S.151–164.
- Grantham JJ: Autosomal Dominant Polycystic Kidney Disease. N Engl J Med 2008; Band 359: S.1477–1485.
- Steinacker JM, Liu Y, Reißnecker S: Abbruchkriterien bei der Ergometrie. Deutsche Zeitschrift für Sportmedizin. 2002; Jahrgang 53, Nr.7+8; S.228–229.
- Walz G, Budde K, Mannaa M, et al.: Everolimus in Patients with Autosomal Dominant Polycystic Kidney Disease. N Engl J Med 2010; Band 363: S.830–840.
- Hildebrandt F, Benzing Th, Katsanis N: Ciliopathies. N Engl J Med 2010; Band 364: S.1533-43.
- Pei Y, Obaji J, Dupuis A, et al.: Unified Criteria for Ultrasonographic Diagnosis of ADPKD. J Am Soc Nephrol 2008; Band 20: S.205–212.
- Haag-Weber M: Peritonealdialyse. Der Nephrologe. 2012; Band 7: S.515–526.
- Grantham JJ, Torres VE, Chapman AB, et al.: Volume Progression in Polycystic Kidney Disease. N Engl J Med 2006; Band 354: S.2122–2130.
- Torres VE, Chapman AB, Devuyst O: Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease. N Engl J Med 2012; Band 367: S.2407–2418.
- Orphanet; The portal for rare diseases and orphan drugs. [Online] 08. August 2013. [Zitat vom: 08. August 2013.] www.orpha.net.
- Ravine D, Gibson RN, Walker RG et al.: Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; Band 343: S.824–827.
Datum: 03.02.2014
Quelle: Wehrmedizinische Monatsschrift 2014/1